Decoding the Innate Immune Response to Exogenous mRNA Delivery: Mechanisms, Challenges, and Therapeutic Optimization
This article provides a comprehensive analysis of the innate immune system's reaction to exogenously delivered mRNA, a cornerstone of modern vaccinology and therapeutics.
Decoding the Innate Immune Response to Exogenous mRNA Delivery: Mechanisms, Challenges, and Therapeutic Optimization
Abstract
This article provides a comprehensive analysis of the innate immune system's reaction to exogenously delivered mRNA, a cornerstone of modern vaccinology and therapeutics. Tailored for researchers and drug development professionals, it explores the fundamental mechanisms of immune sensing by pattern recognition receptors (PRRs), the dual adjuvant roles of mRNA and lipid nanoparticles (LNPs), and the critical balance between immunogenicity and efficacy. The scope extends to advanced delivery strategies, computational and experimental optimization techniques to mitigate unwanted immune activation, and comparative analyses of immune outcomes. By synthesizing recent findings, this review aims to guide the rational design of safer and more effective mRNA-based therapies.
The First Line of Defense: Fundamental Mechanisms of Innate Immune Sensing for mRNA
The innate immune system employs a sophisticated array of pattern recognition receptors (PRRs) to detect invading pathogens through the recognition of conserved molecular signatures. In the context of RNA detection, particularly relevant to exogenous mRNA delivery, three principal receptor families play crucial roles: Toll-like receptors (TLRs), Retinoic acid-inducible gene I (RIG-I), and Melanoma differentiation-associated gene 5 (MDA5). These systems provide complementary surveillance mechanisms that sense RNA in different cellular compartments and initiate signaling cascades leading to interferon and inflammatory cytokine production. Understanding the precise mechanisms of these pathways is fundamental to advancing therapeutic applications, including mRNA vaccine development, where balancing immunogenicity with reactogenicity remains a critical challenge [1] [2].
Classification and Cellular Localization of RNA-Sensing PRRs
RNA-sensing PRRs are strategically localized within cells to provide comprehensive surveillance of both extracellular and intracellular spaces. Membrane-bound TLRs reside in endosomal compartments, while cytosolic RIG-I-like receptors (RLRs) survey the intracellular environment for foreign RNA. This compartmentalization enables the immune system to discriminate between self and non-self RNA based on both molecular patterns and location [1] [3].
Table 1: Classification and Localization of Major RNA-Sensing PRRs
| Receptor Family | Specific Receptors | Cellular Localization | Primary RNA Ligands |
|---|---|---|---|
| Toll-like Receptors (TLRs) | TLR3, TLR7, TLR8 | Endosomal membranes | dsRNA (TLR3), ssRNA (TLR7/8) |
| RIG-I-like Receptors (RLRs) | RIG-I, MDA5, LGP2 | Cytosol | Short dsRNA with 5'-triphosphate (RIG-I), long dsRNA (MDA5) |
| Other Cytosolic Sensors | PKR, OAS | Cytosol | dsRNA |
The strategic localization of these receptors prevents inappropriate activation by self-RNA while ensuring rapid detection of viral invaders. Endosomal TLRs primarily survey internalized material, making them particularly relevant to mRNA vaccine delivery via lipid nanoparticles, which are trafficked through endosomal pathways. In contrast, cytosolic RLRs detect replication intermediates of RNA viruses, providing a crucial second line of defense against intracellular pathogens [2] [3].
Toll-like Receptors: Endosomal RNA Sensing
Structural Features and Ligand Recognition
Toll-like receptors are transmembrane glycoproteins characterized by extracellular leucine-rich repeat (LRR) domains that mediate ligand recognition and intracellular Toll/interleukin-1 receptor (TIR) domains that initiate signaling. TLR3 recognizes double-stranded RNA (dsRNA), while TLR7 and TLR8 sense single-stranded RNA (ssRNA) motifs, particularly those rich in guanosine and uridine [1] [3]. These receptors are predominantly expressed in immune cells such as plasmacytoid dendritic cells (pDCs), conventional dendritic cells, and macrophages, with TLR7 showing particularly high expression in pDCs, which are specialized for massive interferon production upon viral detection [3].
In unstimulated cells, TLRs reside in the endoplasmic reticulum and translocate to endosomes upon cellular activation. This translocation depends on the UNC93B1 trafficking protein and the molecular chaperone gp96 [3]. The endosomal localization confines TLR recognition to internalized ligands, preventing constant activation by self-RNA and making this system particularly relevant for mRNA vaccines delivered via lipid nanoparticles that traffic through the endosomal pathway [2].
Signaling Pathways and Downstream Effects
Upon ligand binding, TLRs dimerize and undergo conformational changes that bring their intracellular TIR domains into proximity, enabling the recruitment of adapter proteins. TLR3 uniquely signals through the TRIF adapter, while TLR7 and TLR8 utilize the MyD88 adapter [1] [3]. This initiates a signaling cascade that culminates in the activation of three major signaling nodes: mitogen-activated protein kinases (MAPKs), interferon regulatory factors (IRFs), and nuclear factor kappa B (NF-κB).
Table 2: TLR Signaling Components and Functions
| Signaling Component | TLR Association | Function |
|---|---|---|
| MyD88 | TLR7, TLR8 | Primary adaptor; recruits IRAK proteins |
| TRIF | TLR3 | Primary adaptor; activates TBK1 and IRF3 |
| IRAK4, IRAK1 | TLR7, TLR8 | Serine-threonine kinases; phosphorylate downstream targets |
| TRAF6 | TLR7, TLR8 | E3 ubiquitin ligase; activates TAK1 complex |
| IRF7 | TLR7, TLR8 | Master regulator of type I IFN genes |
| IRF3 | TLR3 | Activates IFN-β gene expression |
The specific transcription factors activated determine the cytokine profile produced. IRF3 and IRF7 drive type I interferon production, while NF-κB and AP-1 activate pro-inflammatory cytokine genes. This results in the production of type I interferons (IFN-α/β), which establish an antiviral state in neighboring cells, and inflammatory cytokines (IL-6, TNF, IL-1β) that recruit and activate additional immune cells [3]. The outcome is the induction of antiviral effector mechanisms and the initiation of adaptive immune responses through enhanced antigen presentation and co-stimulation.
Figure 1: TLR Signaling Pathways for RNA Sensing
RIG-I-like Receptors: Cytosolic RNA Sensing
Structural Organization and Activation Mechanisms
The RIG-I-like receptor family comprises three members: RIG-I, MDA5, and LGP2. These receptors share a common domain architecture consisting of two N-terminal caspase activation and recruitment domains (CARDs), a central DExD/H-box RNA helicase domain (comprising Hel1, Hel2, and Hel2i subdomains), and a C-terminal domain (CTD). RIG-I and MDA5 both contain N-terminal CARD domains that initiate signaling, while LGP2 lacks CARD domains and functions primarily as a regulator of RIG-I and MDA5 signaling [4] [5] [6].
In the absence of viral RNA, RIG-I exists in an auto-repressed conformation where the CARD domains are sequestered through interactions with the Hel2i domain. This strategic arrangement prevents unintended signaling in the absence of genuine viral infection. Upon encounter with appropriate RNA ligands, RIG-I undergoes major conformational changes that release the CARD domains, enabling downstream signaling [4] [5]. In contrast, MDA5 does not sequester its CARD domains in the inactive state but instead relies on filament formation along RNA ligands for activation [4].
Molecular Basis of RNA Discrimination
RIG-I and MDA5 employ distinct strategies for RNA recognition that enable them to detect different types of viral infections:
RIG-I specifically recognizes short double-stranded RNA (typically <300 base pairs) containing a 5'-triphosphate (5'-ppp) moiety and a blunt-ended double-stranded structure. This molecular signature is characteristic of many RNA virus genomes and replication intermediates, but is absent from host cytoplasmic RNA, which either possesses a 5'-cap structure (mRNA) or is monophosphorylated (processed RNA) [4] [5] [6]. The RIG-I CTD directly engages the 5'-triphosphate group, while the helicase domain wraps around the RNA backbone, forming extensive contacts primarily with the ribose 2'-OH groups rather than the bases, ensuring sequence-independent recognition of double-stranded structure [5].
MDA5 differs fundamentally in its recognition mechanism, specifically sensing long double-stranded RNA (typically >1,000 base pairs) through cooperative assembly of helical filaments along the RNA backbone. Unlike RIG-I, MDA5 shows no specificity for 5'-end structures and instead binds internally to long dsRNA molecules. The ATP-sensitive nature of MDA5 filaments provides a mechanism for length discrimination, as stable filament formation requires sufficiently long RNA substrates [4] [6].
Table 3: Ligand Specificity of RIG-I-like Receptors
| Receptor | RNA Ligand Features | Representative Viruses Detected |
|---|---|---|
| RIG-I | Short dsRNA (<300 bp) with 5'-triphosphate and blunt ends | Influenza virus, vesicular stomatitis virus, paramyxoviruses |
| MDA5 | Long dsRNA (>1,000 bp) without end specificity | Picornaviruses, noroviruses, coronaviruses |
| LGP2 | Various dsRNA structures | Regulatory functions for RIG-I and MDA5 |
Signal Transduction through MAVS
Upon RNA binding and activation, both RIG-I and MDA5 undergo ubiquitin-dependent and ubiquitin-independent oligomerization, enabling them to interact with the central adaptor protein mitochondrial antiviral signaling protein (MAVS, also known as IPS-1, VISA, or Cardif) on mitochondrial and peroxisomal membranes [4] [7]. This interaction triggers MAVS polymerization into prion-like aggregates that serve as scaffolding platforms for recruiting downstream signaling components.
The MAVS signalosome activates two kinase complexes: the IKK complex (which activates NF-κB) and the TBK1/IKKε complex (which phosphorylates IRF3 and IRF7). This leads to the induction of type I interferons (IFN-α/β), type III interferons (IFN-λ), and pro-inflammatory cytokines [4] [7]. The subcellular localization of MAVS signaling influences the specific response, with mitochondrial MAVS promoting type I IFN and inflammatory cytokine production, while peroxisomal MAVS induces early type III IFN expression [4].
Figure 2: RIG-I-like Receptor Signaling Pathway
Experimental Approaches for Studying RNA-Sensing Pathways
Ligand Recognition and Binding Assays
Investigating RNA-PRR interactions requires specialized methodologies to elucidate binding specificity and affinity:
Electrophoretic Mobility Shift Assays (EMSAs) remain a fundamental technique for analyzing direct RNA-protein interactions. Purified recombinant RIG-I or MDA5 helicase domains are incubated with radiolabeled or fluorescently-labeled RNA ligands, and protein-RNA complexes are resolved via non-denaturing polyacrylamide gel electrophoresis. Shifted migration indicates complex formation [4] [5]. For RIG-I studies, specific 5'-triphosphate-containing RNA duplexes (10-19 bp) are employed, while MDA5 binding requires long dsRNA structures (>1,000 bp) such as poly(I:C) [4] [5].
Surface Plasmon Resonance (SPR) and Isothermal Titration Calorimetry (ITC) provide quantitative data on binding kinetics and thermodynamics. SPR measures real-time association and dissociation rates when RNA analytes flow over immobilized PRR proteins, while ITC directly measures the heat changes during binding interactions, providing values for binding stoichiometry, affinity (Kd), and thermodynamic parameters [5].
X-ray Crystallography and Cryo-electron Microscopy have been instrumental in determining high-resolution structures of PRR-RNA complexes. Crystallographic analyses of RIG-I bound to short dsRNA ligands revealed the molecular details of 5'-triphosphate recognition and the conformational changes accompanying activation [4] [5]. Cryo-EM studies have visualized MDA5 filament formation on long dsRNA, providing insights into its cooperative assembly mechanism [4].
Functional Signaling Assays
Determining the functional consequences of PRR activation employs both in vitro and in vivo approaches:
Reporter gene assays are widely used to quantify pathway activation. Cells are transfected with PRR expression plasmids along with reporter constructs containing interferon-stimulated response elements (ISRE) or NF-κB binding sites driving firefly luciferase expression. Activation is measured as luciferase activity following stimulation with specific RNA ligands [5] [7].
Gene knockout and knockdown models establish non-redundant functions in antiviral defense. Mouse embryonic fibroblasts (MEFs) from RIG-I⁻/⁻ and MDA5⁻/⁻ mice show distinct vulnerabilities to different RNA viruses, demonstrating the specialized roles of these receptors [6] [7]. RNA interference in human cell lines provides complementary loss-of-function data.
Cytokine and interferon measurements quantify physiological outputs of pathway activation. ELISA and multiplex bead-based arrays measure type I interferon (IFN-α/β) and pro-inflammatory cytokine (IL-6, TNF, IL-1β) production from primary immune cells or cell lines following stimulation with specific RNA ligands [2] [7].
The Scientist's Toolkit: Key Research Reagents
Table 4: Essential Reagents for Studying RNA-Sensing PRRs
| Reagent Category | Specific Examples | Research Applications |
|---|---|---|
| Specific Agonists | Poly(I:C) (long), 5'-ppp RNA, Imidazoquinolines (R848) | Selective activation of MDA5 (long poly(I:C)), RIG-I (5'-ppp RNA), and TLR7/8 (R848) |
| Inhibitors | BX795 (TBK1/IKKε inhibitor), RIG-I/NN (RIG-I inhibitor), Chloroquine (endosomal acidification blocker) | Pathway dissection and validation of specific signaling components |
| Antibodies | Phospho-IRF3, Phospho-TBK1, Total RIG-I/MDA5, MAVS | Detection of protein expression, phosphorylation, and activation status via Western blot, immunofluorescence |
| Cell Lines | HEK293-TLR reporter cells, RIG-I⁻/⁻ and MDA5⁻/⁻ MEFs, Human pDC and macrophage models | Pathway-specific signaling analysis, loss-of-function studies, primary cell responses |
| Animal Models | MAVS⁻/⁻ mice, MyD88⁻/⁻ TRIF⁻/⁻ mice, Conditional knockout models | In vivo validation of pathway functions, therapeutic development |
Implications for Exogenous mRNA Delivery Systems
The development of mRNA-based therapeutics, particularly vaccines, requires careful consideration of PRR activation. Unmodified in vitro transcribed mRNA is highly inflammatory due to recognition by multiple RNA sensors, which initially posed a significant challenge for therapeutic applications [2]. Several strategies have been implemented to modulate this immunogenicity:
Nucleoside modification represents a cornerstone technology for reducing unwanted immune activation. Replacement of uridine with pseudouridine (Ψ) or N1-methylpseudouridine (m1Ψ) allows mRNA to evade or mitigate detection by most innate immune sensors, resulting in reduced inflammation and greatly improved protein translation [2]. This approach mimics naturally occurring modified nucleosides in host RNA, providing a mechanism for self versus non-self discrimination.
Purification techniques remove double-stranded RNA contaminants that are potent RIG-I and MDA5 agonists. HPLC purification and RNase III treatment effectively eliminate dsRNA byproducts generated during in vitro transcription, significantly reducing innate immune activation [2].
Delivery system optimization balances efficient cytosolic delivery with appropriate adjuvant effects. Ionizable lipid nanoparticles (iLNPs) not only facilitate mRNA delivery but also contribute adjuvant activity through mechanisms that are not yet fully understood but may involve specific PRRs or inflammasome activation [2]. The current COVID-19 mRNA vaccines demonstrate that the iLNP component itself induces chemokines and pro-inflammatory cytokines including CCL2, IL-6, and IFN-γ [2].
Understanding and manipulating these PRR interactions enables the rational design of mRNA therapeutics with optimized translation efficiency and controlled immunogenicity, balancing effective adaptive immune induction with acceptable reactogenicity profiles.
The sophisticated network of pattern recognition receptors that detect RNA molecules represents a critical interface between host defense and therapeutic innovation. The compartmentalized nature of TLR versus RIG-I/MDA5 sensing, coupled with their distinct ligand specificities, provides layered protection against diverse RNA viruses while maintaining tolerance to self-RNA. For mRNA vaccine development and other RNA-based therapeutics, precise modulation of these pathways enables the striking of an optimal balance between immunogenicity and reactogenicity. Future research will continue to refine our understanding of regulatory mechanisms, cross-talk between pathways, and cell-type-specific responses, ultimately enabling more precise targeting of these systems for therapeutic benefit.
Lipid nanoparticle (LNP)-encapsulated mRNA vaccines represent a transformative advance in vaccinology, demonstrated most notably by their rapid deployment during the COVID-19 pandemic. These vaccines elicit robust adaptive immune responses through a sophisticated interplay between their two fundamental components: the mRNA molecule encoding the antigenic protein and the LNP delivery vehicle that facilitates intracellular delivery [8] [2]. Despite their clinical success, the precise immunological mechanisms underlying their efficacy are still being elucidated. Current research reveals that both components contribute significantly to innate immune activation, which serves as a critical bridge to adaptive immunity [9] [10]. This technical guide examines the distinct roles of mRNA structure and LNP adjuvanticity in triggering innate immune responses, providing researchers with a comprehensive resource on the fundamental principles governing this innovative vaccine platform.
The mRNA Component: More Than Just Coding Sequence
Structural Elements and Their Immunological Implications
The mRNA in vaccines is not merely a linear coding sequence but a complex molecular architecture with specific structural features that profoundly influence protein expression and immunogenicity. In vitro-transcribed (IVT) mRNA mimics endogenous eukaryotic mRNA, containing five critical regions: the 5' cap, 5' untranslated region (UTR), open reading frame (ORF), 3' UTR, and poly(A) tail [8]. Each component serves distinct functions in translation efficiency, stability, and innate immune recognition.
The 5' cap structure, consisting of 7-methylguanosine linked to the first nucleotide via a triphosphate bridge, plays dual roles in preventing exonuclease degradation and reducing recognition by cytosolic RNA sensors [8]. Importantly, 2'-O-methylation of the first or second nucleotide abrogates detection by pattern recognition receptors (PRRs) that would otherwise trigger antiviral responses. The poly(A) tail (typically 100-150 nucleotides) interacts with poly(A)-binding proteins to circularize the mRNA molecule, enhancing ribosome recruitment and translation initiation while protecting the 5' cap from decapping enzymes [8]. Both the 5' and 3' UTRs regulate mRNA translation, half-life, and subcellular localization, with optimized UTR sequences from highly expressed genes (e.g., α- and β-globin) minimizing mRNA degradation by excluding miRNA-binding sites and AU-rich elements [8].
Table 1: Structural Components of Synthetic mRNA and Their Functions
| mRNA Component | Key Features | Biological Functions | Impact on Immunogenicity |
|---|---|---|---|
| 5' Cap | 7-methylguanosine; 2'-O-methylation | Prevents degradation; enables translation initiation | Reduces recognition by IFIT proteins and RIG-I |
| 5' UTR | Optimized sequences (e.g., α-globin) | Regulates ribosome scanning and loading | Minimizes secondary structures that activate PRRs |
| Coding Region | Nucleoside modifications (m1Ψ); codon optimization | Encodes antigen; determines translation efficiency | Modified nucleosides prevent TLR7/8 activation |
| 3' UTR | Stabilizing elements; AU-rich region depletion | Controls mRNA stability and half-life | Eliminates motifs that promote rapid degradation |
| Poly(A) Tail | 100-150 nucleotides; encoded in template | Enhances translation; protects from degradation | Optimal length balances expression and reduced immunogenicity |
Nucleoside Modifications and Purification Strategies
A critical breakthrough in mRNA vaccine development came from understanding how innate immune sensors discriminate between self and non-self RNA. Karikó and Weissman discovered that replacing uridine with naturally occurring derivatives (pseudouridine, N1-methylpseudouridine [m1Ψ]) allows mRNA to evade detection by numerous innate immune sensors, particularly endosomal TLR7/TLR8 [8] [2]. This nucleoside modification reduces inflammatory signaling and markedly enhances protein expression by preventing translational inhibition through PKR activation and OAS-mediated RNA degradation pathways.
Removal of double-stranded RNA (dsRNA) contaminants during manufacturing represents another crucial optimization. These dsRNA byproducts, generated during in vitro transcription, are potent ligands for multiple intracellular sensors including TLR3, RIG-I, MDA5, and PKR [2]. Stringent purification methods, such as cellulose-based purification or RNase III digestion, efficiently remove dsRNA contaminants, further reducing unintended immune activation and improving translational capacity [9] [2].
The LNP Component: Delivery System and Intrinsic Adjuvant
Composition and Structural Properties
LNPs are sophisticated delivery vehicles that protect mRNA from degradation and facilitate its cellular uptake and endosomal release. Clinically approved LNPs typically comprise four lipid components, each serving distinct structural and functional roles [10] [8] [2].
The ionizable lipid (e.g., ALC-0315 in Comirnaty, SM-102 in Spikevax) is the most critical component, with a pKa of approximately 6.0-6.8. This property enables the LNP to remain neutral at physiological pH but acquire positive charge in acidifying endosomes, facilitating endosomal membrane disruption and mRNA release into the cytosol [2]. Phospholipids (e.g., DSPC) and cholesterol contribute to LNP structural integrity and facilitate endosomal escape, while PEGylated lipids enhance colloidal stability, prevent particle aggregation, and prolong circulation half-life by reducing nonspecific interactions with plasma proteins and cellular components [10] [8].
Table 2: Components of Lipid Nanoparticles and Their Functions
| LNP Component | Example Molecules | Molar Ratio | Primary Function |
|---|---|---|---|
| Ionizable Lipid | ALC-0315, SM-102 | ~40-50% | mRNA encapsulation; endosomal escape; adjuvant activity |
| Phospholipid | DSPC | ~10% | Structural stability; membrane fusion facilitation |
| Cholesterol | Natural cholesterol | ~38-40% | Membrane integrity; fluidity modulation |
| PEGylated Lipid | DMG-PEG, ALC-0159 | ~1.5-2% | Colloidal stability; reduced opsonization; pharmacokinetics |
Innate Immune Activation by LNPs
While initially developed primarily as delivery vehicles, LNPs are now recognized as potent intrinsic adjuvants that significantly contribute to vaccine immunogenicity [10] [2]. Multiple studies demonstrate that empty LNPs (devoid of mRNA) can induce robust innate immune activation, characterized by production of proinflammatory cytokines (IL-6, TNF-α, IL-1β) and chemokines (CCL2, CCL3, CXCL10) [9] [2].
This adjuvant activity appears particularly dependent on the ionizable lipid component, which can activate various innate immune pathways. LNP administration triggers rapid recruitment and activation of innate immune cells, including monocytes and dendritic cells, to the injection site and draining lymph nodes [9] [11]. This creates a pro-inflammatory microenvironment that supports subsequent adaptive immune responses, enhancing antigen-specific antibody production and T cell activation [10] [2].
Integrated Immune Activation Mechanisms
Synergistic Actions of mRNA and LNP Components
While each component exhibits intrinsic immunostimulatory properties, their combination in LNP-mRNA vaccines creates a synergistic system that optimally engages both innate and adaptive immunity. Recent research employing comparative approaches with empty LNPs, non-coding mRNA, and complete vaccines has begun to delineate the specific contributions of each element [9] [11].
The mRNA component, even when nucleoside-modified, appears essential for inducing type I interferon (IFN-α/β) responses, particularly in migratory dendritic cells and injection-site fibroblasts [9] [11]. Conversely, the LNP component primarily drives proinflammatory cytokine production and immune cell recruitment through distinct signaling pathways [11] [2]. This division of labor creates a comprehensive innate immune milieu that effectively primes subsequent antigen-specific responses.
Spatiotemporal Dynamics of Immune Activation
The immune response to LNP-mRNA vaccines follows a carefully orchestrated spatiotemporal sequence beginning at the injection site. Within hours of administration, LNPs facilitate mRNA uptake by local cells, particularly stromal fibroblasts and immune cells [11]. These cells then initiate distinct response programs: fibroblasts produce IFN-β in response to intracellular mRNA sensing, while the LNP component triggers inflammatory cytokine production [11].
This initial response recruits and activates antigen-presenting cells, which subsequently migrate to draining lymph nodes to prime naive T cells and initiate germinal center reactions [10] [11]. The type I IFN response induced by the mRNA component plays a particularly crucial role in shaping adaptive immunity, enhancing dendritic cell maturation and cross-priming of CD8+ T cells [9] [11].
Immune Activation Pathway
Experimental Approaches and Methodologies
Comparative Vaccine Formulations
To dissect the specific contributions of mRNA and LNP components to immune activation, researchers have developed sophisticated experimental approaches using controlled vaccine formulations [9] [11]. These typically include: (1) complete LNP-mRNA vaccines, (2) empty LNPs (devoid of mRNA), (3) LNPs encapsulating non-coding mRNA sequences, and (4) LNPs with different mRNA payloads encoding distinct antigens.
In one representative study [9], LNP formulations were prepared using a microfluidic mixer (NanoAssemblr) with lipid components mixed at a molar ratio of 40:47.5:10.5:2 (ionizable lipid:cholesterol:DSPC:DMG-PEG) and mRNA dissolved in citrate buffer (pH 4.5). The resulting particles exhibited hydrodynamic sizes of 60-70 nm with low polydispersity indices (0.11-0.23) and high encapsulation efficiency (>93%), ensuring consistent vaccine characteristics across experimental groups [9].
Immune Monitoring Techniques
Comprehensive immune profiling employs multiple complementary techniques to capture both innate and adaptive responses. Single-cell RNA sequencing of injection site tissues and draining lymph nodes has proven particularly valuable for identifying responding cell populations and their transcriptional programs [11]. In one such approach, researchers profiled 83,094 single cells from vaccine injection sites, revealing distinct response axes: PC1 representing stromal pro-inflammatory responses (LNP-driven) and PC2 representing type I IFN responses (mRNA-driven) [11].
Flow cytometry-based immunophenotyping enables quantification of immune cell recruitment and activation, while cytokine profiling (ELISA, multiplex assays) characterizes the soluble inflammatory milieu. Assessment of adaptive immunity includes plaque reduction neutralization tests for antibody function, ELISpot for antigen-specific T cell responses, and intracellular cytokine staining for detailed T cell characterization [9] [11].
Experimental Workflow for Immune Profiling
The Scientist's Toolkit: Key Research Reagents and Materials
Table 3: Essential Research Reagents for mRNA Vaccine Immunology Studies
| Category | Specific Reagents | Application/Function | Key References |
|---|---|---|---|
| mRNA Constructs | Nucleoside-modified mRNA (m1Ψ); Non-coding mRNA; Cellulose-purified mRNA | Component contribution studies; Control for antigen-specific effects | [9] [8] |
| LNP Components | Ionizable lipids (ALC-0315, SM-102); DSPC; Cholesterol; DMG-PEG | LNP formulation; Adjuvant mechanism studies | [9] [2] |
| Animal Models | C57BL/6J mice; IFNAR-/- mice; BALB/c mice | In vivo vaccine efficacy; Immune mechanism studies | [9] [11] |
| Immunological Reagents | Anti-IFNAR blocking antibodies; Flow cytometry antibodies; Cytokine detection kits | Immune pathway inhibition; Immune cell phenotyping; Cytokine quantification | [9] [11] |
| Analytical Instruments | NanoAssemblr; Dynamic light scattering; Zetasizer; scRNA-seq platform | LNP formulation; Particle characterization; Immune profiling | [9] [11] |
The remarkable efficacy of LNP-mRNA vaccines stems from the sophisticated interplay between their two core components: the mRNA molecule, which encodes the antigen while simultaneously modulating innate immune recognition through its structural features; and the LNP delivery system, which provides both intracellular delivery and potent intrinsic adjuvant activity. Understanding the distinct and synergistic roles of these components provides valuable insights for optimizing current vaccine platforms and designing next-generation mRNA-based therapeutics. Future research directions include engineering novel ionizable lipids with improved safety profiles, optimizing mRNA structural elements for cell-type specific expression, and fine-tuning the balance between immunogenicity and reactogenicity for specific clinical applications.
The efficacy of mRNA vaccines hinges on a carefully choreographed innate immune response at the injection site. This whitepaper delineates the distinct yet synergistic roles of fibroblasts, dendritic cells (DCs), and monocytes in the initial hours post-vaccination. Groundbreaking single-cell transcriptomic analyses reveal that fibroblasts are primary targets for vaccine mRNA, initiating a critical type I interferon (IFN-β) response. This IFN-β, in turn, activates migratory DCs and shapes subsequent cellular immunity. Monocytes and other innate cells are recruited, contributing to a pro-inflammatory milieu. Understanding this cellular cascade provides a mechanistic framework for optimizing next-generation mRNA vaccines and therapeutics, a central theme in exogenous mRNA delivery research.
The advent of lipid nanoparticle (LNP)-formulated mRNA vaccines has revolutionized vaccinology. While their ability to induce potent adaptive immunity is well-established, the initial innate immune events at the injection site that orchestrate this response are only now being unraveled [11]. This early phase is critical, as it sets the stage for the quality and magnitude of the antigen-specific response. The mRNA vaccine platform is unique in its built-in adjuvanticity, provided by both the LNP and the mRNA itself, eliminating the need for exogenous adjuvants [12]. Within the complex tissue environment of the injection site, stromal and immune cells act as the first responders. Recent research provides a detailed map of these interactions, identifying fibroblasts, dendritic cells, and monocytes as key cellular orchestrators that detect vaccine components, initiate signaling cascades, and prime the immune system for a robust and targeted defense [11] [13]. This whitepaper synthesizes recent findings to provide an in-depth technical guide for researchers and drug development professionals.
Distinct Roles of Key Cellular Orchestrators
Comprehensive single-cell transcriptome profiling of mRNA vaccine injection sites has identified three major cell types with specialized functions in the early immune response.
Fibroblasts: The Initial Sentinels and IFN-β Hubs
Musculoskeletal fibroblasts are among the first and most significant cells to encounter and respond to the mRNA vaccine.
- mRNA Uptake: Lineage tracking of delivered mRNA reveals that injection-site fibroblasts are highly enriched with the vaccine mRNA, serving as a primary reservoir for antigen translation [11].
- Specific IFN-β Production: A pivotal discovery is that fibroblasts express IFN-β specifically in response to the mRNA component of the vaccine, not the LNP alone. This positions them as the initiators of a key antiviral signaling pathway [11] [14].
- Downstream Signaling: The IFN-β produced by fibroblasts does not act in isolation. It acts in a paracrine manner to induce a distinct population of migratory Dendritic Cells highly expressing IFN-stimulated genes (mDC_ISGs) at the injection site and draining lymph nodes (dLNs) [11].
Migratory Dendritic Cells (mDCs): The Interferon-Responsive Bridges to Adaptive Immunity
Dendritic cells are potent antigen-presenting cells essential for T-cell priming. A specific subset is critically modulated by the fibroblast-derived signal.
- mDC_ISG Induction: The mRNA-LNP vaccine, but not LNP alone, induces a unique population of migratory DCs characterized by high expression of interferon-stimulated genes (ISGs) such as
Isg15,Oasl1, andIfit3[11]. - Dependence on IFN-β: The emergence of mDC_ISGs is directly dependent on IFN-β signaling. Blocking IFN-β signaling at the injection site significantly decreases this DC population and the subsequent cellular immune response [11].
- Role in Immunity: These activated DCs are equipped to travel to draining lymph nodes, where they provide the three signals necessary for T-cell priming: antigen presentation (Signal 1), costimulation (Signal 2 via CD80/CD86), and polarizing cytokines (Signal 3) [12].
Monocytes and the Pro-Inflammatory Axis
The LNP component of the vaccine drives a robust pro-inflammatory response, largely mediated by innate immune cells like monocytes.
- LNP-Driven Recruitment: Injection of either empty LNP or mRNA-LNP leads to a prominent increase in monocyte, neutrophil, and CD8 T-cell populations at the injection site within 16 hours [11] [13].
- Inflammatory Cytokine Production: This cellular recruitment is driven by the LNP-induced expression of pro-inflammatory cytokines and chemokines, such as IL-6, TNF, and CCL2, predominantly from stromal cells [11].
- Systemic Activation: In humans, mRNA vaccination increases circulating classical and intermediate inflammatory monocytes and enhances their IFN-γ production, indicating systemic activation [13].
Table 1: Key Cellular Orchestrators at the mRNA Vaccine Injection Site
| Cell Type | Primary Stimulus | Key Transcriptional Signature/Output | Functional Consequence |
|---|---|---|---|
| Fibroblast | mRNA component | IFN-β production | Initiates type I IFN response; induces mDC_ISGs |
| Migratory DC (mDC_ISG) | IFN-β (from fibroblasts) | ISGs (Isg15, Oasl1, Ifit3) |
Bridges innate & adaptive immunity; T-cell priming |
| Monocytes / Myeloid Cells | LNP component | Pro-inflammatory cytokines (IL6, TNF, CCL2) |
Recruitment of innate immune cells; inflammation |
Quantitative Data on Early Immune Responses
The orchestrated response peaks at a specific timeframe and involves a precise sequence of events.
Temporal Dynamics of the Injection Site Response
Single-cell RNA sequencing time-course experiments show that transcriptional responses at the injection site culminate at approximately 16 hours post-injection [11]. Principal component analysis of differentially expressed genes reveals two major, independent axes of response:
- PC1 (Stromal Inflammation): Driven by the LNP component, this axis features pro-inflammatory genes and is active in both empty-LNP and mRNA-LNP injected tissues.
- PC2 (Antiviral/Interferon): Driven by the mRNA component, this axis is highly specific to mRNA-LNP injected samples and is characterized by type I IFN and antiviral response genes in mDCs [11].
Cellular Tropism of mRNA Vaccine
Quantification of spike mRNA-positive cells at the injection site 2 hours post-injection reveals distinct cellular tropism.
Table 2: Cellular Tropism of Delivered mRNA at the Injection Site (2 Hours Post-Injection)
| Cell Type | Relative Enrichment of Spike mRNA |
|---|---|
| Fibroblasts | High |
| Endothelial Cells | High |
| Pericytes | High |
| Myeloid Cells (e.g., Monocytes, DCs) | Moderate |
| Lymphoid Cells (T cells, B cells) | Low |
The detection rate of spike mRNA decreases over time, likely due to the degradation of mRNA molecules [11].
Experimental Workflow and Methodologies
The following diagram illustrates the key experimental workflow used to dissect the innate immune response at the injection site, from challenge to single-cell analysis.
Detailed Experimental Protocol
1. In Vivo Challenge and Sample Collection:
- Model: Female BALB/c mice.
- Immunization: Intramuscular injection with saline (PBS), empty LNP, or nucleoside-modified mRNA-LNP encoding SARS-CoV-2 spike glycoprotein. Prime and boost shots are administered with a 3-week interval [11].
- Time Points: The injection site (anterior thigh muscle) and draining lymph nodes are resected at serial time points from 2 to 40 hours post-injection for single-cell analysis [11].
- Validation: Blood and spleen samples are collected 2 weeks post-boost to confirm vaccine efficacy using Plaque Reduction Neutralization Test (PRNT) and IFN-γ ELISpot assays [11].
2. Single-Cell RNA Sequencing Preparation:
- Tissue Dissociation: Resected muscle tissues are subjected to both mechanical and chemical digestion to generate high-viability single-cell suspensions [11].
- Library Construction: Single-cell suspensions are used to construct barcoded scRNA-seq libraries using standard platforms (e.g., 10x Genomics). This captures the transcriptome of tens of thousands of individual cells [11].
3. Immunophenotyping by Flow Cytometry (for Validation Studies):
- Blood Collection: Human whole blood is collected in EDTA vacutainers from vaccinated individuals at multiple time points [13].
- Cell Staining: Blood is incubated with fluorochrome-labeled monoclonal antibodies targeting surface markers (e.g., for monocytes: CD14, CD16; for NK cells: CD3, CD56, CD16) [13].
- Intracellular Cytokine Staining: Isolated PBMCs are stimulated ex vivo with recombinant SARS-CoV-2 spike protein (S1 subunit) in the presence of brefeldin A. Cells are then fixed, permeabilized, and stained intracellularly for cytokines (e.g., IFN-γ, TNF) and cytotoxic molecules (perforin, granzyme) [13].
- Acquisition and Analysis: A flow cytometer (e.g., Beckman-Coulter GALLIOS) is used to acquire at least 20,000 events per sample, with data analyzed using specialized software (e.g., Kaluza) [13].
Signaling Pathways and Cellular Cross-Talk
The innate immune response to mRNA vaccination is a cascade initiated by cellular detection of vaccine components, leading to a coordinated response. The following diagram summarizes the key signaling pathways and cellular interactions.
Mechanistic Insights from Interferon Blockade
The critical role of the fibroblast-IFN-β-mDC axis was confirmed through loss-of-function experiments:
- Intervention: IFN-β signaling was blocked locally at the injection site [11].
- Observation: This intervention led to a significant decrease in mDC_ISGs and, crucially, a substantial reduction in mRNA vaccine-induced antigen-specific cellular immune responses [11].
- Conclusion: These findings demonstrate that the IFN-β pathway is not merely correlative but is mechanistically required for optimal T-cell priming by mRNA vaccines.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Investigating mRNA Vaccine Innate Immunity
| Reagent / Tool | Function / Target | Application in Research |
|---|---|---|
| Nucleoside-modified mRNA-LNP | Delivers antigen-encoding mRNA; provides dual adjuvant effect. | In vivo challenge to model vaccine response; core component of experimental systems [11]. |
| Empty LNP (no mRNA) | Control for the adjuvant effect of the lipid nanoparticle. | Disentangles immunogenicity of mRNA from LNP in comparative studies [11]. |
| Anti-IFNAR1 blocking antibody | Blocks the type I interferon receptor (IFNAR). | Used in vivo to inhibit IFN-β signaling and validate its functional role in immune induction [11]. |
| Recombinant Spike Protein (S1 subunit) | SARS-CoV-2 antigen for ex vivo stimulation. | Used in intracellular cytokine staining (ICS) to measure antigen-specific T-cell and innate immune responses [13]. |
| Fluorochrome-conjugated Antibodies | Cell surface and intracellular protein markers. | Flow cytometry phenotyping (e.g., CD14, CD16 for monocytes; CD11c, MHC-II for DCs) and ICS (IFN-γ, TNF) [13]. |
| Single-Cell RNA-Seq Kits (e.g., 10x Genomics) | High-throughput transcriptomic profiling of individual cells. | Unbiased identification of cell types, transcriptional states, and differential gene expression at the injection site [11]. |
The initial immune response to mRNA vaccination is a precisely coordinated event orchestrated by a consortium of cells at the injection site. Fibroblasts act as pivotal sentinels, detecting the mRNA component and launching a critical IFN-β signal. This cytokine dictates the differentiation of a specialized subset of migratory DCs (mDC_ISGs), which are essential for bridging the innate and adaptive arms of immunity. Concurrently, the LNP component drives a pro-inflammatory program that recruits and activates monocytes and other myeloid cells. This detailed mechanistic understanding of the early innate immune cascade provides a robust scientific foundation for the rational design of next-generation mRNA vaccines, with potential strategies including the modulation of IFN responses or the targeted delivery of mRNA to specific cell types to enhance efficacy and reduce reactogenicity.
Type I interferons (IFN-α/β) represent a critical cornerstone of the innate immune response to exogenous mRNA delivery, orchestrating a complex cytokine cascade that profoundly influences both host defense and therapeutic efficacy. This whitepaper delineates the molecular mechanisms of IFN-α/β signaling initiated by mRNA-loaded lipid nanoparticles (LNPs), detailing the pattern recognition receptors involved, the subsequent JAK/STAT signaling pathway, and the resulting transcriptional program. Within the context of mRNA vaccine research, we examine the dual role of IFN-α/β in enhancing antigen presentation and adaptive immunity while potentially limiting antigen translation. This guide provides a technical resource for researchers and drug development professionals, featuring quantitative data analyses, standardized experimental protocols, and essential research tools to advance the field of innate immunology and mRNA therapeutics.
Type I interferons, primarily IFN-α and IFN-β, are pleiotropic cytokines that constitute the host's first line of defense against viral pathogens and are central players in the immune response to exogenous mRNA. They are produced by nearly all nucleated cells upon detection of foreign nucleic acids and signal through a common receptor, IFNAR (IFN-α/β receptor), to establish an antiviral state [15] [10]. The IFNAR receptor is composed of two subunits, IFNAR1 and IFNAR2, and its engagement triggers the canonical JAK/STAT signaling pathway, leading to the transcription of hundreds of interferon-stimulated genes (ISGs) [16] [10]. These ISGs execute diverse antiviral functions, ranging from inhibiting viral translation and replication to promoting apoptosis of infected cells.
The induction and function of IFN-α/β create a fundamental paradox in the context of mRNA vaccine and therapeutic development. On one hand, their signaling is crucial for activating dendritic cells, promoting T-cell responses, and generating robust humoral immunity [17] [9]. On the other hand, the IFN-α/β response can inhibit the translation of the encoded antigen, potentially reducing the yield of the desired immunogen and contributing to vaccine reactogenicity [9] [10]. A precise understanding of this delicate balance is therefore imperative for designing next-generation mRNA platforms with optimized efficacy and safety profiles.
Quantitative Data Analysis of IFN-α/β Signaling
The following tables summarize key quantitative findings from recent studies on IFN-α/β signaling in the context of immune activation and mRNA vaccine responses.
Table 1: Quantifiable Effects of IFN-α/β Signaling in Preclinical and Clinical Contexts
| Observation | Quantitative Measure | Experimental Model | Source |
|---|---|---|---|
| Enhanced survival with mRNA vaccination + ICI | HRadj = 0.51 (95% CI: 0.37-0.71); Median OS: 20.6 vs 37.3 months | NSCLC patients | [18] |
| IFNAR blockade enhances adaptive immunity | Increased frequencies of antigen-specific CD8+ T cells; Elevated antigen-specific antibodies | Murine model | [9] |
| Species difference in IFN-α response | 8 to 16-fold stronger response levels in mice vs. humans | QSP model (In vivo/In silico) | [16] |
| In vitro vs. In vivo drug effect | In vitro effect overestimates in vivo response by a factor of two | QSP model (In silico) | [16] |
| FRET efficiency in STAT5 activation | Up to 12% FRET efficiency upon IL-2 stimulation | Live cell biosensor (STATeLight) | [19] |
Table 2: Key Interferon-Stimulated Genes (ISGs) and Their Functions
| ISG | Function | Experimental/Clinical Context |
|---|---|---|
| Mx2 | Dynamin-like GTPase with antiviral activity against a wide range of viruses | Used as a pharmacodynamic biomarker for IFN-α activity in mouse hepatocytes [16] |
| ISG15 | Ubiquitin-like protein that conjugates to target proteins (ISGylation) | Downregulated in COVID-19 patients with functional variants of TLR7 [17] |
| PKR (EIF2AK2) | Serine/threonine-protein kinase that phosphorylates eIF2α, inhibiting translation | Implicated in the reduction of immunogen protein synthesis following IFN response to mRNA [10] |
Signaling Pathways and Molecular Mechanisms
Initiation: Recognition of Exogenous mRNA
The cascade begins with the cellular detection of exogenous mRNA, primarily by endosomal and cytosolic pattern recognition receptors (PRRs). Toll-like receptor 7 (TLR7) within endosomes recognizes single-stranded RNA (ssRNA) and is particularly crucial for the sex-skewed severity observed in COVID-19, with loss-of-function variants increasing the risk of life-threatening disease [17] [20]. Concurrently, the cytosolic RNA sensors RIG-I and MDA5 can detect mRNA, signaling through the mitochondrial antiviral-signaling protein (MAVS) to activate transcription factors like IRF3 and NF-κB [20] [10]. The lipid nanoparticles (LNPs) used for mRNA delivery further contribute to this activation by serving as both carriers and adjuvants, potentially engaging additional innate immune pathways [10].
The JAK/STAT Signaling Cascade
The binding of IFN-α/β to the IFNAR receptor initiates the canonical JAK/STAT signaling pathway. This association activates the receptor-associated Janus kinases (JAKs), TYK2 and JAK1, which subsequently phosphorylate tyrosine residues on the intracellular tail of IFNAR. These phospho-tyrosines serve as docking sites for the SH2 domains of STAT1 and STAT2 proteins. Upon recruitment, STAT1 and STAT2 are themselves phosphorylated by JAKs, leading to their dissociation from the receptor, dimerization, and association with a third protein, IRF9. This complex, known as ISGF3 (Interferon-Stimulated Gene Factor 3), translocates to the nucleus where it binds to specific DNA sequences called Interferon-Stimulated Response Elements (ISREs) in the promoters of ISGs, thereby initiating their transcription [16] [15] [10].
Diagram 1: The canonical JAK/STAT signaling pathway activated by IFN-α/β binding.
Biological Outcomes and the Dual-Edged Sword
The transcriptional program launched by ISGF3 has multifaceted consequences. It establishes an antiviral state in the cell, inhibiting various stages of pathogen replication. Furthermore, IFN-α/β signaling acts as a powerful bridge between innate and adaptive immunity. It enhances the maturation and antigen-presenting capacity of dendritic cells, promotes the differentiation of T cells, and supports B cell antibody production [17] [18]. However, this potent immune activation is a double-edged sword. The same IFN-α/β signaling can lead to the phosphorylation of PKR, which in turn phosphorylates eukaryotic initiation factor 2α (eIF2α), globally inhibiting translation and thereby reducing the production of the encoded antigen from the therapeutic mRNA [9] [10]. This negative feedback loop highlights the critical balance that must be struck in mRNA vaccine design.
Experimental Protocols and Methodologies
Protocol: Measuring IFN-α/β-Mediated JAK/STAT Activation Using a Genetically Encoded Biosensor
The STATeLight biosensor enables real-time, continuous monitoring of STAT activation in live cells via FLIM-FRET (Fluorescence Lifetime Imaging Microscopy - Förster Resonance Energy Transfer) [19].
- Biosensor Design: The biosensor is engineered by tagging STAT5A monomers with a FRET pair—mNeonGreen (mNG, donor) and mScarlet-I (mSC-I, acceptor)—at their C-termini, directly following the SH2 domain. This positioning ensures a detectable change in FRET efficiency upon cytokine-induced conformational change from antiparallel to parallel dimers.
- Cell Preparation and Transfection:
- Culture HEK-Blue IL-2 cells (or other relevant cell line) in appropriate medium.
- Transfect cells with the STATeLight5A plasmid construct using a standard method (e.g., lipofection, electroporation).
- Allow 24-48 hours for expression of the biosensor.
- Stimulation and Imaging:
- Stimulate cells with interleukin-2 (IL-2) at a concentration of 10-100 ng/mL to activate the JAK/STAT pathway.
- Perform FLIM-FRET imaging on a confocal microscope equipped with a time-correlated single-photon counting (TCSPC) module.
- Excite the mNG donor fluorophore with a pulsed laser (e.g., 488 nm) and measure its fluorescence lifetime.
- Data Analysis:
- A decrease in the fluorescence lifetime of the donor (mNG) indicates increased FRET efficiency, signifying STAT5A dimerization and activation.
- Quantify FRET efficiency using the formula:
E = 1 - (τ_DA / τ_D), whereτ_DAis the donor lifetime in the presence of the acceptor, andτ_Dis the donor lifetime alone.
Protocol: In Vivo Evaluation of LNP-mRNA Immunogenicity and IFNAR Dependence
This protocol assesses the innate and adaptive immune response to LNP-mRNA vaccines and the specific role of IFN-α/β signaling [9].
- Vaccine Preparation:
- Use nucleoside-modified mRNA (e.g., complete N1-methyl-pseudouridine substitution) purified to remove dsRNA contaminants.
- Encapsulate mRNA in LNPs composed of ionizable lipid, cholesterol, DSPC, and DMG-PEG. Include control groups with "empty" LNPs (lacking mRNA) and PBS.
- Animal Model and Immunization:
- Use age-matched, 6-8 week old female C57BL/6J (wild-type) and IFNAR-/- mice.
- Administer vaccine (e.g., 5 μg LNP-mRNA) via intramuscular injection into the hind legs.
- IFNAR Blocking:
- To dissect the role of IFNAR signaling, inject wild-type mice intraperitoneally with 2.5 mg of anti-IFNAR monoclonal antibodies 24 hours prior to and 24 hours post immunization.
- Immune Response Analysis:
- Innate Immunity (Day 1-2): Analyze dendritic cell activation, monocyte recruitment to draining lymph nodes, and systemic cytokine levels (e.g., via ELISA or multiplex immunoassay).
- Adaptive Immunity (Weeks 1-4): Measure antigen-specific CD8+ T cell frequencies (via flow cytometry with tetramer staining) and antigen-specific antibody titers (via ELISA) after the final immunization.
Diagram 2: Experimental workflow for evaluating LNP-mRNA immunogenicity and IFNAR dependence in vivo.
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for Investigating IFN-α/β Signaling in mRNA Research
| Research Reagent | Function/Application | Example Use Case |
|---|---|---|
| Anti-IFNAR monoclonal antibody | Blocks the type I interferon receptor, allowing dissection of IFNAR-specific effects. | Used in vivo to demonstrate that transient IFNAR inhibition enhances adaptive immune responses to LNP-mRNA vaccines [9]. |
| STATeLight Biosensor | Genetically encoded FRET-based biosensor for real-time monitoring of STAT activation in live cells. | Enabled direct, continuous detection of STAT5A conformational changes and dimerization upon cytokine stimulation [19]. |
| IFN-α/β (Murine & Human) | Recombinant cytokine proteins for exogenous stimulation and standard curve generation in assays. | Used in vitro to stimulate primary hepatocytes and establish dose-response relationships for ISG induction [16]. |
| JAK/STAT Pathway Inhibitors (e.g., Deucravacitinib) | Small molecule inhibitors targeting key nodes in the JAK/STAT signaling cascade. | Used to pharmacologically validate the role of specific kinases in the IFN-induced signaling pathway [9]. |
| ELISA/Multiplex Assay Kits | Quantify cytokine and chemokine protein levels (e.g., IFN-α, IFN-β, IP-10, IL-6) in cell culture supernatants or serum. | Essential for measuring the innate immune cytokine profile following LNP-mRNA administration in both pre-clinical models and human studies [9] [18]. |
| LNP Formulations (mRNA-loaded vs. Empty) | Delivery vehicle for mRNA; empty LNPs serve as a control to separate the immunogenicity of the carrier from the payload. | Critical for demonstrating that the mRNA component, rather than the LNP alone, is essential for robust IFNAR-dependent innate activation [9] [10]. |
Implications for mRNA Vaccine and Therapeutic Development
The understanding of the IFN-α/β cascade has direct and profound implications for the design of mRNA vaccines and therapeutics. Strategies to modulate this response are actively being pursued. These include the use of nucleoside modifications (e.g., N1-methylpseudouridine) and highly purified mRNA to minimize unwanted PRR activation, thereby reducing innate signaling and enhancing antigen translation [9] [20]. Alternatively, the timed modulation of IFNAR signaling presents a promising approach. Transient inhibition of IFNAR, as demonstrated in murine models, can enhance adaptive immune responses by preventing the IFN-mediated inhibition of antigen translation, without completely abolishing the beneficial adjuvant effects of the cytokine [9].
Beyond infectious diseases, the immunomodulatory power of LNP-mRNA-induced IFN-α/β is being harnessed in oncology. Recent groundbreaking research has shown that SARS-CoV-2 mRNA vaccines, through their induction of a type I interferon surge, can reset the tumor microenvironment and sensitize immunologically "cold" tumors to immune checkpoint blockade (ICI). This effect was associated with significantly improved overall survival in patients with non-small cell lung cancer and melanoma who received an mRNA vaccine shortly before or during ICI treatment [18]. This repurposing of clinically available mRNA vaccines as general immune modulators opens a new frontier in cancer immunotherapy.
The revolutionary success of messenger RNA (mRNA) vaccines against COVID-19 represents a paradigm shift in vaccinology, showcasing the critical importance of understanding the intricate interplay between innate and adaptive immune systems. These lipid nanoparticle (LNP)-encapsulated, nucleoside-modified mRNA vaccines function not merely as antigen delivery systems but as sophisticated immunomodulatory platforms that orchestrate a precise immune cascade beginning within hours of administration [2] [21]. The core principle underlying their efficacy lies in their capacity to be sensed by the innate immune system, which in turn provides the necessary instructional signals to shape qualitatively and quantitatively superior adaptive immune responses encompassing neutralizing antibodies, helper T cells, and cytotoxic T lymphocytes [21] [22].
This technical guide examines the fundamental mechanisms through which exogenous mRNA delivery platforms activate innate immunity and how this activation bridges to the establishment of protective adaptive immunity. We focus specifically on the context of nucleoside-modified mRNA-LNP vaccines—the platform used in the licensed Pfizer/BioNTech and Moderna COVID-19 vaccines—while providing detailed methodological approaches for investigating these immune pathways. A comprehensive understanding of these processes is essential for researchers aiming to optimize current mRNA vaccine platforms or develop novel mRNA-based therapeutics for infectious diseases, cancer, and other applications.
Innate Immune Sensing of Exogenous mRNA
Pattern Recognition Receptors and Their Ligands
The innate immune system detects exogenous mRNA through multiple pattern recognition receptors (PRRs) that recognize molecular signatures as foreign. These PRRs are strategically located in various cellular compartments to detect both extracellular and cytosolic RNA encounters.
Table 1: Major Innate Immune Receptors Sensing mRNA Vaccine Components
| Receptor | Location | Ligand | Signaling Pathway | Primary Cell Types |
|---|---|---|---|---|
| TLR3 | Endosome | dsRNA | TRIF → IRF3/NF-κB | DCs, Macrophages |
| TLR7/8 | Endosome | ssRNA | MyD88 → IRF7/NF-κB | pDCs, Macrophages |
| RIG-I | Cytosol | dsRNA, 5'ppp RNA | MAVS → IRF3/NF-κB | Fibroblasts, Epithelial cells |
| MDA5 | Cytosol | long dsRNA | MAVS → IRF3/NF-κB | Various cell types |
| PKR | Cytosol | dsRNA | eIF2α phosphorylation | Various cell types |
| NLRP3 | Cytosol | Multiple | Inflammasome → IL-1β | Monocytes, Macrophages |
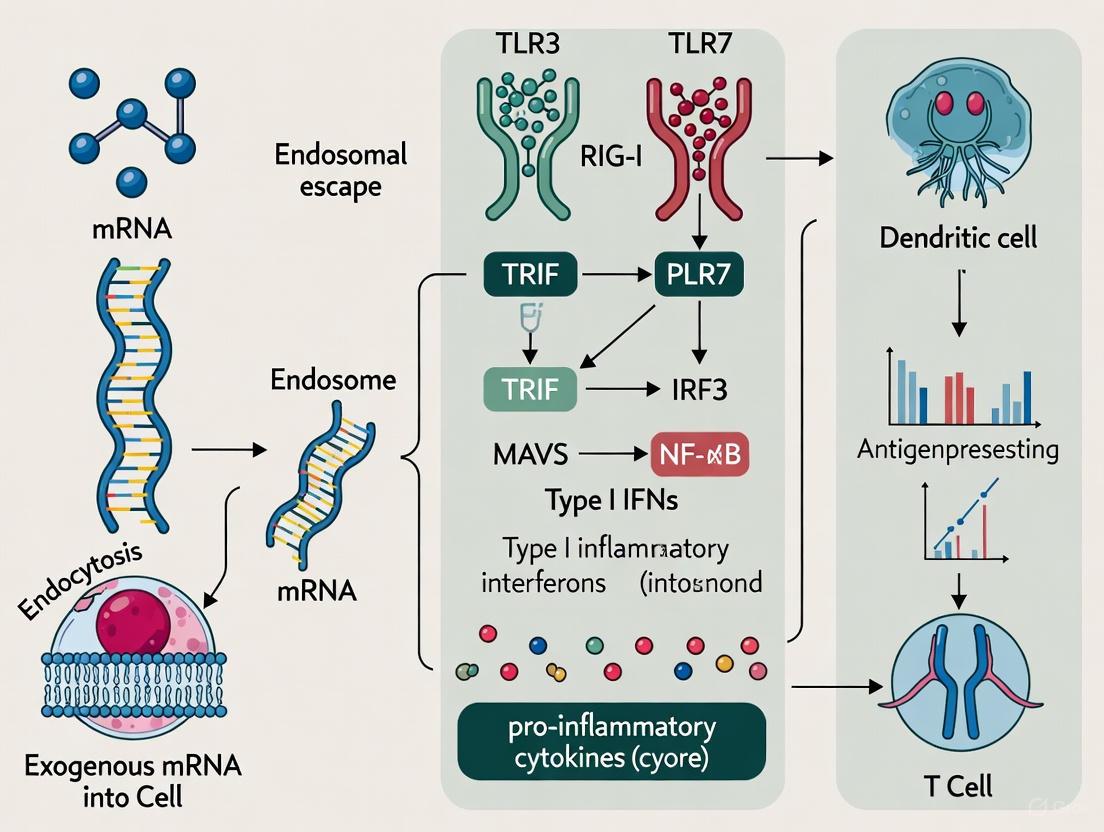
Upon intramuscular administration, mRNA-LNPs are taken up by local cells at the injection site, including myocytes, fibroblasts, and resident immune cells such as dendritic cells (DCs) and macrophages [11] [22]. The LNP shell protects the mRNA cargo and facilitates cellular entry primarily through endocytosis. Within the acidifying endosome, the ionizable lipids undergo protonation, enabling endosomal escape and release of mRNA into the cytosol where translation occurs [2] [22]. Both the mRNA molecule itself and the LNP delivery vehicle contribute to innate immune activation through distinct but complementary mechanisms.
mRNA Component Recognition
In vitro transcribed (IVT) mRNA possesses several structural features that can trigger PRR activation. Double-stranded RNA (dsRNA) contaminants generated during the transcription process are potent agonists for TLR3, RIG-I, MDA5, and protein kinase R (PKR) [2] [22]. Even single-stranded mRNA can be sensed by TLR7/8 in endosomal compartments and potentially by RIG-I in the cytosol, particularly if it contains specific sequence motifs or lacks appropriate modifications [22].
Nucleoside modification represents a crucial technological advancement that mitigates excessive innate immune activation while enhancing protein expression. Replacement of uridine with naturally occurring derivatives such as pseudouridine (Ψ) or N1-methylpseudouridine (m1Ψ) enables mRNA to evade detection by many innate sensors, thereby reducing inflammatory signaling and preventing translational inhibition [2] [23]. This modification, coupled with sophisticated purification methods to remove dsRNA contaminants, allows modern mRNA vaccines to achieve the delicate balance between sufficient innate activation for adjuvanticity and controlled inflammation for safety and high antigen expression [2] [22].
LNP Component Recognition
While early research emphasized the immuno-silent nature of nucleoside-modified mRNA, recent evidence demonstrates that the LNP carrier itself functions as a potent adjuvant [2] [9]. The ionizable lipid component—SM-102 in the Moderna vaccine and ALC-0315 in the Pfizer/BioNTech vaccine—is particularly critical for this adjuvanticity [2] [22]. Although the precise sensing mechanisms for LNPs remain incompletely characterized, emerging data suggest they may activate inflammatory pathways, including possibly the NLRP3 inflammasome, and induce cytokine production in a manner dependent on their chemical structure [2] [22].
Table 2: Key LNP Components and Their Immunological Functions
| Component | Function | Impact on Innate Immunity |
|---|---|---|
| Ionizable lipid (e.g., ALC-0315, SM-102) | Enables endosomal escape, mRNA release | Primary driver of LNP adjuvanticity; induces IL-6, cytokine production |
| PEG-lipid | Stabilizes nanoparticle, reduces opsonization | Modulates protein adsorption, affects immunogenicity |
| Cholesterol | Stabilizes LNP structure | May influence cellular uptake and endosomal escape |
| Phospholipid (e.g., DSPC) | Structural support | Generally non-inflammatory |
From Sensing to Signaling: Intracellular Pathways and Early Immune Responses
Signal Transduction and Transcriptional Activation
PRR engagement by mRNA vaccine components triggers intricate intracellular signaling cascades that culminate in the production of type I interferons (IFN-α/β), proinflammatory cytokines, and chemokines. Endosomal TLR activation primarily signals through either the MyD88 adaptor (TLR7/8) or TRIF adaptor (TLR3), while cytosolic RIG-I-like receptors signal through the mitochondrial antiviral signaling protein (MAVS) [22]. These signaling pathways converge on the activation of transcription factors including IRF3, IRF7, and NF-κB, which translocate to the nucleus and induce the expression of interferon-stimulated genes (ISGs) and inflammatory mediators [11] [22].
Single-cell transcriptomic analyses of mRNA vaccine injection sites in mouse models have revealed that these early innate responses follow distinct temporal and cellular patterns. Stromal cells (fibroblasts, endothelial cells) exhibit strong proinflammatory responses characterized by the production of cytokines such as IL-6, TNF, and CCL2, primarily driven by the LNP component [11]. Conversely, migratory dendritic cells specifically upregulate type I interferon response genes, including ISG15, OASL1, and IFIT3, in reaction to the mRNA component [11].
Diagram 1: Innate immune signaling pathway initiated by mRNA-LNP vaccines, showing the sequence from cellular uptake to immune activation.
Early Cellular Responses at Injection Site and Draining Lymph Nodes
The cytokine and chemokine milieu established at the injection site orchestrates the recruitment and activation of innate immune cells. Within hours of mRNA-LNP vaccination, neutrophils, monocytes, and inflammatory DCs infiltrate the muscle tissue [11]. Notably, even empty LNPs (without mRNA) can induce significant innate cellular recruitment, though the combination of LNP and mRNA generates qualitatively distinct responses [11] [24].
A pivotal cell population in the early immune response to mRNA vaccines is migratory dendritic cells expressing interferon-stimulated genes (mDC_ISGs). These specialized antigen-presenting cells are specifically induced by the mRNA component of the vaccine and exhibit enhanced capacity for antigen presentation and T cell priming [11]. Through tracking the fate of administered mRNA, researchers have identified fibroblasts at the injection site as key early responders that are highly enriched with delivered mRNA and produce IFN-β specifically in response to the mRNA component [11].
The early innate response rapidly extends to the draining lymph nodes, where activated dendritic cells present vaccine antigen to naïve T cells and initiate the germinal center reaction essential for B cell maturation and antibody production [9] [22]. Within 24 hours of vaccination, significant activation of dendritic cells and monocytes is observable in the draining lymph nodes, creating a microenvironment conducive to the development of adaptive immunity [9].
Bridging to Adaptive Immunity
Orchestrating Humoral and Cellular Responses
The innate immune activation triggered by mRNA-LNP vaccination directly shapes the quality, magnitude, and persistence of adaptive immune responses through multiple mechanisms. The cytokines and chemokines produced during the innate phase promote dendritic cell maturation, enhance antigen presentation, and provide crucial co-stimulatory signals for T cell activation [21] [22].
Type I interferons play a particularly important role in bridging innate and adaptive immunity. IFN-α/β signaling enhances cross-priming of CD8+ T cells, promotes B cell class switching, and supports the development of T follicular helper cells that are essential for germinal center formation [11] [9]. However, the timing and magnitude of type I interferon signaling require precise regulation, as excessive or prolonged signaling can potentially suppress antigen expression and impair adaptive immune responses [9].
Table 3: Key Innate Immune Signals and Their Impact on Adaptive Immunity
| Innate Signal | Source | Adaptive Immune Effect | Molecular Mechanism |
|---|---|---|---|
| Type I IFNs (IFN-α/β) | Stromal cells, DCs | Enhances CD8+ T cell cross-priming, Th1 polarization | ISG expression, MHC-I upregulation |
| IL-6 | Myeloid cells | Promotes TFH differentiation, antibody production | STAT3 activation |
| Inflammatory cytokines (TNF, IL-1β) | Multiple innate cells | Enhances DC maturation, T cell activation | NF-κB signaling, costimulatory molecule expression |
| Chemokines (CCL2, CCL3, CCL4) | Stromal and immune cells | Recruits monocytes, T cells, DCs | Chemokine receptor engagement |
Spatiotemporal Regulation of Immune Responses
The effectiveness of mRNA vaccines in generating robust adaptive immunity depends critically on the spatiotemporal coordination of antigen expression and innate immune activation. Ideally, antigen expression reaches sufficient levels before innate sensing mechanisms trigger an antiviral state that could potentially suppress further translation [2] [9]. The LNP delivery system helps coordinate this timing by controlling the release kinetics of mRNA and simultaneously providing adjuvant signals that create an immunogenic microenvironment.
Research comparing different mRNA vaccine formulations has demonstrated that combining antigens can enhance immunogenicity through modulation of innate immune responses. For instance, mice vaccinated with both spike (S) and nucleocapsid (N) mRNA exhibited heightened innate immune activation with increased IL-6 and MCP-1 production, alongside enhanced germinal center reactions and T cell responses compared to single-antigen vaccination [24] [25]. This synergistic effect underscores how vaccine formulation can be optimized to leverage innate-adaptive immune crosstalk.
Experimental Approaches for Investigating mRNA Vaccine Immunity
Comprehensive Methodologies for Immune Profiling
mRNA-LNP Preparation and Characterization: Research-grade mRNA vaccines can be synthesized using T7 RNA polymerase-based in vitro transcription with complete substitution of uridine with N1-methylpseudouridine (m1Ψ) [9] [24]. The mRNA should be purified using cellulose-based methods or HPLC to remove immunostimulatory dsRNA contaminants [9]. LNPs are typically formulated using microfluidic mixing with ionizable lipids (e.g., ALC-0315), phospholipids (DSPC), cholesterol, and PEG-lipids (DMG-PEG2000) at defined molar ratios [9] [24]. Critical quality control measurements include nanoparticle size (60-80 nm), polydispersity index (<0.2), encapsulation efficiency (>90%), and endotoxin levels [9].
Innate Immune Profiling: To evaluate early innate responses, mice are immunized intramuscularly with mRNA-LNPs (1-5 μg dose), and samples are collected at 8-24 hours post-injection [24]. Serum cytokines (IL-6, MCP-1, IFN-α) can be quantified using multiplex bead-based assays [24]. Single-cell transcriptomics of injection site tissues provides comprehensive mapping of cellular responses and identification of key responder cell populations [11]. Flow cytometric immunophenotyping of draining lymph nodes assesses activation markers on dendritic cells (CD80, CD86, MHC-II) and natural killer cells [24] [25].
Adaptive Immune Assessment: For evaluation of adaptive immunity, mice receive prime-boost vaccinations 3 weeks apart, with analysis 1-2 weeks post-boost [24]. Antigen-specific antibody titers (IgG, subtypes) are measured by ELISA, while neutralizing capacity is assessed using pseudovirus or live virus neutralization assays [9]. Antigen-specific T cell responses are evaluated by intracellular cytokine staining (IFN-γ, TNF, IL-2) after peptide stimulation, MHC multimer staining, or ELISpot assays [9] [24]. Germinal center reactions can be analyzed by flow cytometry of lymph node or spleen cells for T follicular helper cells (CXCR5+PD-1+Bcl-6+) and germinal center B cells (GL-7+Fas+) [22].
Diagram 2: Experimental workflow for comprehensive immune profiling of mRNA vaccines, from preparation to innate and adaptive immune analysis.
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents for Investigating mRNA Vaccine Immunity
| Reagent/Category | Specific Examples | Research Application | Technical Notes |
|---|---|---|---|
| mRNA Constructs | Nucleoside-modified mRNA (m1Ψ), unmodified mRNA, non-coding RNA | Component-specific immune analysis, control conditions | Cellulose purification critical for reducing dsRNA contaminants |
| LNP Components | Ionizable lipids (ALC-0315, SM-102), DSPC, Cholesterol, DMG-PEG2000 | Formulation optimization, adjuvant studies | Molar ratios significantly impact immunogenicity |
| Animal Models | C57BL/6, BALB/c mice, IFNAR-/- mice, humanized mice | In vivo vaccine efficacy, mechanistic studies | IFNAR-/- mice essential for type I IFN pathway investigation |
| Cytokine Detection | LEGENDplex panels, ELISA kits, Luminex | Innate immune profiling, correlates of immunogenicity | Early timepoints (8-24h) critical for innate cytokine measurement |
| Immune Cell Analysis | Flow cytometry antibodies (CD45, CD3, CD19, CD11c, MHC-II), intracellular cytokine staining | Cellular immune responses, activation status | Comprehensive panels for innate and adaptive cell subsets |
| Pathway Modulators | Anti-IFNAR blocking antibodies, Deucravacitinib (TYK2 inhibitor) | Mechanistic studies of signaling pathways | Timing critical for pathway blockade experiments |
Concluding Perspectives and Future Directions
The intricate interplay between innate sensing and adaptive immunity represents both the challenge and promise of mRNA vaccine technology. The dual role of innate activation—as necessary adjuvant and potential barrier to antigen expression—underscores the importance of precisely engineered mRNA and delivery systems. Future research directions should focus on elucidating the specific PRRs responsible for LNP recognition, developing strategies for spatial and temporal control of innate activation, and designing next-generation mRNA constructs with tunable immunostimulatory properties.
As the field advances, the fundamental principle remains clear: the bridge from innate sensing to adaptive signaling is the cornerstone of mRNA vaccine efficacy. A deeper mechanistic understanding of this immunological dialogue will enable researchers to optimize this transformative platform for broader applications while maintaining the favorable safety profile that has made mRNA vaccines a revolutionary tool in preventive medicine.
Engineering the Message: Methodologies for Controlling Immune Activation
The advent of nucleoside-modified messenger RNA (mRNA) represents a pivotal advancement in the field of nucleic acid therapeutics, enabling the development of effective vaccines and treatments by overcoming major immunological hurdles. A cornerstone of this technology is the incorporation of N1-methylpseudouridine (m1Ψ), a modified nucleoside that allows synthetic mRNA to evade detection by the innate immune system. This evasion is critical for enhancing the translation efficiency and safety of mRNA-based drugs. This whitepaper details the molecular mechanisms by which m1Ψ modulates immune sensing, summarizes key quantitative findings from preclinical and clinical studies, and provides a toolkit of standard experimental protocols. Framed within the broader context of innate immune response to exogenous mRNA delivery, this review underscores how strategic nucleoside modification has unlocked the therapeutic potential of mRNA platforms.
The innate immune system is equipped with a sophisticated network of pattern recognition receptors (PRRs) that vigilantly scan for foreign molecular patterns, a defense mechanism crucial for host survival [20]. Exogenously delivered mRNA, a key component of modern therapeutic platforms, is inherently recognized as a pathogen-associated molecular pattern (PAMP) by these receptors [2] [26]. This recognition triggers potent antiviral defense pathways, leading to the suppression of protein translation and the induction of inflammatory cytokines, which collectively can undermine the efficacy of mRNA drugs and cause undesirable adverse effects [2] [23].
The seminal discovery that certain naturally occurring nucleoside modifications could dampen this immune activation paved the way for viable mRNA therapeutics. Among these, the replacement of uridine with pseudouridine (Ψ) and its derivative m1Ψ proved to be particularly effective [26] [23]. This breakthrough addressed a fundamental obstacle: how to deliver functional mRNA without triggering an overwhelming innate immune response. The subsequent success of m1Ψ-modified mRNA in COVID-19 vaccines validated this approach and highlighted the importance of understanding the intricate relationship between mRNA chemistry and immune sensing [2] [27]. This paper explores how m1Ψ serves as a molecular stealth technology, enabling the safe and efficient use of mRNA in biomedical applications.
Molecular Mechanisms of Immune Evasion
The immune-evasive properties of m1Ψ are mediated through its ability to alter the molecular signature of synthetic mRNA, thereby reducing its engagement with key PRRs. The following diagram illustrates the primary sensing pathways for unmodified mRNA and how m1Ψ modification intervenes.
Key Immune Sensing Pathways and m1Ψ Intervention
Toll-like Receptor 7/8 (TLR7/8): Residing in endosomal membranes, TLR7/8 are specialized in sensing single-stranded RNA (ssRNA), particularly sequences rich in uridine [20]. The incorporation of m1Ψ fundamentally alters the molecular structure of the mRNA, preventing its recognition by these receptors. This is a primary mechanism by which m1Ψ-modified mRNA avoids triggering a robust type I interferon (IFN) response [2] [26].
RIG-I-like Receptors (RLRs): Cytosolic sensors, including RIG-I and MDA5, detect viral RNA. RIG-I is activated by RNA features such as 5'-triphosphate ends. While the 5' cap structure of synthetic mRNA is a primary method to evade RIG-I, the use of m1Ψ provides an additional layer of immune silencing by further reducing the immunogenic profile of the mRNA molecule [20] [27]. Studies with the BNT162b2 vaccine have shown that the CD8+ T cell response it induces is dependent on MDA5 signaling, but not on TLR signaling, highlighting the complex and nuanced role of different PRR pathways in mRNA vaccine immunogenicity [27].
The synergistic effect of combining m1Ψ modification with other design features, such as a cap1 structure and optimized untranslated regions (UTRs), creates an mRNA molecule that the host cell's machinery translates efficiently without mounting a significant antiviral defense [2] [23].
Quantitative Experimental Evidence
The efficacy of m1Ψ is demonstrated by quantifiable improvements in protein expression and reductions in immune activation across various experimental models. The data below summarize key findings from in vitro and in vivo studies.
Table 1: Impact of m1Ψ Modification on Protein Expression and Immune Activation In Vitro
| Cell Type | mRNA Construct | Protein Expression vs. Unmodified | Key Immune Markers | Reference |
|---|---|---|---|---|
| Primary Human Myoblasts (HSKM) | Influenza HA (cKK-E10 LNP) | Significantly higher | N/A | [28] |
| Primary Human Dendritic Cells (hDCs) | Influenza HA (cKK-E10 LNP) | Significantly higher | N/A | [28] |
| RAW264.7 Macrophages | EGFP mRNA | Equivalent expression, 8-fold higher GFP+ cells | Decreased IFN response | [29] |
| HSKM Cells | Global Translation (Puromycin Assay) | ~40-46% higher than unmodified | N/A | [28] |
Table 2: In Vivo Immune Responses to m1Ψ-Modified mRNA Vaccines
| Model System | Vaccine / Construct | Reported Findings | Key Immune Readouts | Reference |
|---|---|---|---|---|
| Mouse Model | BNT162b2 (m1Ψ) | Potent antibody & T cell responses | High IFN-γ post-boost; MDA5-dependent CD8+ T cells | [27] |
| Non-Human Primates | Influenza HA (m1Ψ LNP) | Enhanced functional antibody titers | Strong humoral and cellular immunity | [28] |
| Human Clinical Trial | BNT162b2 & mRNA-1273 | ~95% efficacy against COVID-19 | Robust neutralizing antibodies & TH1-biased T cells | [2] [30] |
The data consistently show that m1Ψ modification enhances translational output, in part by mitigating global translational repression often induced by unmodified mRNA [28]. Furthermore, while m1Ψ significantly reduces innate immune activation, it does not completely abolish it; the lipid nanoparticle (LNP) carrier itself can act as an adjuvant, contributing to a desirable level of immune stimulation for vaccine applications [2] [27].
Essential Protocols for Research and Development
This section outlines core methodologies for evaluating the performance and immunogenicity of m1Ψ-modified mRNA, providing a framework for standard laboratory experiments.
Protocol: Assessing Innate Immune Activation via ISRE Reporter Assay
The IFN-stimulated response element (ISRE) reporter assay is a robust method for quantifying the overall IFN response triggered by mRNA transfection.
- Cell Preparation: Culture ISRE-reporter cells (e.g., RAW-Lucia ISG murine macrophages) according to standard protocols [29].
- MRNA Transfection: Transfect cells with a defined dose (e.g., 100 ng-1 µg) of experimental mRNA (unmodified AUGC, m1Ψ-modified, etc.) using a commercial lipofection reagent. Include a positive control (e.g., unmodified mRNA) and a negative control (e.g., buffer only).
- Incubation: Incubate the cells for 16-24 hours to allow for immune activation and reporter secretion.
- Detection and Quantification: Harvest cell culture supernatant. Quantify luciferase activity (or secreted alkaline phosphatase, SEAP) using a luminometer and appropriate detection reagents. Normalize results to the positive control to determine the percentage reduction in immune activation conferred by m1Ψ [29].
Protocol: Evaluating Protein Expression by Flow Cytometry
This protocol measures the efficiency of antigen production in immune cells, a critical parameter for vaccine development.
- Cell Transfection: Transfect primary human dendritic cells or macrophages with mRNA-LNP formulations encoding a model antigen like enhanced green fluorescent protein (EGFP) or influenza hemagglutinin (HA) [28].
- Incubation: Allow cells to express the encoded protein for 18-24 hours.
- Cell Staining: Harvest cells and stain with a fluorescently labeled antibody specific for the encoded antigen (e.g., anti-HA) or, in the case of EGFP, analyze directly.
- Flow Cytometric Analysis: Analyze the cells using a flow cytometer. The percentage of antigen-positive cells and the mean fluorescence intensity (MFI) provide quantitative measures of translation efficiency. m1Ψ-modified mRNA typically results in a higher percentage of positive cells and a greater MFI compared to unmodified mRNA [28] [29].
Successful research into nucleoside-modified mRNA requires a suite of specialized reagents and delivery systems.
Table 3: Essential Research Reagents for m1Ψ mRNA Studies
| Reagent / Resource | Function and Role | Example Application |
|---|---|---|
| m1Ψ Triphosphate (m1Ψ-UTP) | Chemically modified nucleotide for IVT; replaces UTP to reduce immunogenicity and enhance translation. | Synthesis of low-immunogenicity mRNA for vaccines and therapeutics [26] [23]. |
| Ionizable Lipid Nanoparticles (LNPs) | Delivery vehicle for mRNA; protects mRNA, facilitates cellular uptake and endosomal escape, and provides adjuvant activity. | Formulating mRNA vaccines (e.g., SM-102 in Moderna, ALC-0315 in Pfizer-BioNTech) [2] [31]. |
| ISRE-Reporter Cell Line | Tool for quantifying innate immune activation via IFN pathway signaling. | High-throughput screening of novel mRNA constructs for immunogenicity [29]. |
| Cap Analog (CleanCap) | Co-transcriptional capping to produce Cap 1 structure, which reduces RIG-I sensing and improves translation. | Generating mature, highly translatable mRNA during IVT [26] [20]. |
| T7 RNA Polymerase | Bacteriophage-derived RNA polymerase for high-yield in vitro transcription of mRNA from a DNA template. | Core enzyme for synthesizing research-scale and clinical-grade mRNA [26] [23]. |
Current Challenges and Future Directions
Despite its proven success, the use of m1Ψ is not without limitations. A recent study revealed that m1Ψ modification can, in some contexts, cause ribosomal frameshifting during translation, potentially leading to the production of off-target protein products [26]. While this does not appear to compromise the immune response to vaccines, it raises important considerations for the application of mRNA technology in protein replacement therapies where precise translation is critical.
Future research is focused on exploring next-generation solutions. These include:
- Novel Nucleoside Modifications: Investigating alternative modified purines and pyrimidines. For instance, the ZUGC alphabet, which incorporates the purine analog 2-aminoadenine (Z), has shown promise in evading immune detection through a mechanism distinct from m1Ψ, and may even generate metabolites that suppress TLR7 activation [29].
- Advanced mRNA Architectures: Engineering self-amplifying RNA (saRNA) and circular RNA (circRNA) to achieve longer-lasting protein expression at lower doses, though these present distinct delivery and safety challenges [23].
- Ionizable Lipid Optimization: Designing new ionizable lipids that fine-tune the balance between adjuvant effect (immunogenicity) and tolerability (reactogenicity), as the LNP component synergistically influences the performance of the mRNA payload [2] [28].
The strategic incorporation of N1-methylpseudouridine (m1Ψ) into mRNA therapeutics represents a foundational innovation in the field, directly addressing the central challenge of innate immune recognition. By serving as a molecular decoy, m1Ψ allows exogenous mRNA to bypass key pattern recognition receptors, leading to enhanced protein production and a more favorable safety profile. The quantitative data and standardized protocols outlined in this whitepaper provide a roadmap for researchers to explore and refine this technology. As the field progresses, a deeper understanding of the synergistic effects between nucleoside chemistry, sequence optimization, and advanced delivery systems will undoubtedly unlock the next wave of mRNA-based medicines, extending their application from infectious disease vaccines to cancer immunotherapy, protein replacement, and beyond.
Lipid Nanoparticles (LNPs) have emerged as the leading non-viral delivery platform for messenger RNA (mRNA) therapeutics and vaccines, a fact unequivocally demonstrated by the clinical success of COVID-19 mRNA vaccines. At the heart of the LNP system lies the ionizable lipid, a component that serves two critical, distinct, and often intertwined functions. Primarily, it is the key engineering tool that facilitates the efficient encapsulation, cellular delivery, and endosomal release of mRNA into the cytoplasm for translation [32] [2]. Second, a growing body of evidence identifies the ionizable lipid as a primary driver of the innate immune activation and adjuvanticity of mRNA-LNP formulations [2] [10] [28]. This inherent immunogenicity is a double-edged sword; while it can be harnessed to potentiate robust adaptive immune responses in vaccines, it can also lead to undesired inflammatory side effects and suppress protein expression in non-immunotherapeutic applications [23]. This technical guide delves into the molecular design, mechanisms, and experimental characterization of ionizable lipids, framing their dual role within the context of the innate immune response to exogenous mRNA delivery.
Fundamental Components and Functions of mRNA-LNPs
A typical mRNA-LNP is a multi-component system where each lipid contributes critically to the structure, stability, and function of the nanoparticle. The core composition includes four key constituents, as detailed in the table below.
Table 1: Core Lipid Components of mRNA-LNPs and Their Functions
| Component | Key Function | Molecular Role | Common Examples |
|---|---|---|---|
| Ionizable Lipid | mRNA encapsulation & endosomal escape | Protonatable headgroup; neutral at physiological pH, positively charged in acidic endosomes [2] [10]. | ALC-0315, SM-102, MC3 [24] [2] |
| Phospholipid | Structural integrity & fusogenicity | Stabilizes LNP structure; supports fusion with endosomal membrane [10]. | DSPC [24] [2] |
| Cholesterol | Stability & fluidity | Modulates membrane integrity and fluidity; enhances endosomal escape [10]. | Cholesterol [32] |
| PEG-lipid | Stability & pharmacokinetics | Shields LNP surface, reduces aggregation, controls particle size, and modulates pharmacokinetics [10]. | DMG-PEG2000 [24] |
The ionizable lipid is the most pivotal of these components. Its defining characteristic is a titratable, ionizable amine headgroup with a pKa typically between 6.0 and 6.8 [2] [33]. This specific pKa range is engineered to be neutral or slightly negative at physiological pH (7.4), which reduces nonspecific interactions and prolongs circulation time. However, following cellular uptake via endocytosis, the lipid becomes progressively protonated as the endosome acidifies. This positive charge enables interaction with the anionic endosomal membrane, destabilizing it and facilitating the release of mRNA into the cytosol—a process critical for translational efficiency [32] [10].
Innate Immune Recognition of mRNA-LNPs: A Synergistic Activation
The innate immune system perceives mRNA-LNPs through multiple, synergistic pathways. The immunogenicity of the platform is not solely a property of the mRNA but arises from the combined effects of the mRNA itself and the LNP delivery system, particularly the ionizable lipid [2] [28].
Sensing of mRNA and LNP Components
The innate immune system employs Pattern Recognition Receptors (PRRs) to detect foreign molecular patterns. Exogenous mRNA can be sensed by endosomal Toll-like Receptors (TLR7 and TLR8) and cytosolic sensors like RIG-I and MDA5 [2] [10]. While nucleoside modifications (e.g., N1-methylpseudouridine, m1Ψ) can mitigate RNA sensing, they do not fully ablate it [2] [28].
Critically, the LNP component, and specifically the ionizable lipid, acts as a potent adjuvant. Studies have shown that ionizable lipids can stimulate immune cells to produce pro-inflammatory cytokines such as IL-6 and chemokines like MCP-1 [24] [2]. Empty LNPs (without mRNA) can elicit this response, confirming the lipid itself is immunogenic [24]. The exact PRRs involved in lipid sensing are still being elucidated, with potential roles for TLR2 and TLR4, and other pathways such as the inflammasome [2] [10].
Integrated Signaling Pathway
The following diagram illustrates the synergistic innate immune signaling pathways activated by mRNA-LNPs, involving both the mRNA payload and the ionizable lipid component.
This synergistic immune activation has direct consequences for the adaptive immune response. Cytokines like IL-6 are critical for activating antigen-specific CD4 T follicular helper cells and germinal center B cells, thereby enhancing antibody production [2] [10]. Therefore, the ionizable lipid's adjuvanticity is a key feature that can be rationally designed to tune vaccine efficacy.
Rational Design and Optimization of Ionizable Lipids
The structure of an ionizable lipid dictates its performance, influencing pKa, biodegradability, delivery efficiency, and immunogenicity. Traditional development relied on extensive screening, but new approaches are increasing efficiency.
Structure-Activity Relationship (SAR) and AI-Driven Design
The ionizable lipid structure can be deconstructed into three domains, each with a distinct function:
- Headgroup: Influences pKa and hydrogen bonding with mRNA. A tertiary amine is standard for protonation capability [33].
- Linker: Determines biodegradability and metabolic clearance. Ester linkages are common as they enhance biodegradability and improve safety profiles [2] [33].
- Hydrocarbon Tails: Impact membrane fluidity, packing, and pKa. Saturated alkyl chains enhance stability, while unsaturated chains can increase fusogenicity [33].
Recent advances employ Artificial Intelligence (AI) to navigate this complex design space. Models can predict critical LNP properties like apparent pKa and mRNA delivery efficiency relative to benchmarks like MC3. One study used an AI model to screen nearly 20 million virtual lipids, followed by synthesis and in vivo validation. This approach successfully identified several novel lipids that matched or outperformed SM-102, a lipid used in a licensed COVID-19 vaccine [33]. AI models provide interpretable insights into the molecular substructures that contribute to high efficacy, dramatically accelerating rational design.
Strategies for Modulating Immunogenicity
The immunogenicity of ionizable lipids can be tuned for the application:
- For Vaccines (Pro-Immunogenic): Lipids can be designed to provide strong adjuvant activity. Another strategy is the direct integration of Toll-like receptor (TLR) agonists into the LNP. A 2025 study incorporated a TLR7/8 agonist (AD7/8) into LNPs by partially replacing cholesterol. This formulation (AD03-LNP) significantly enhanced antigen-specific CD8⁺ T cell responses and Th1-skewed antibody (IgG2a) production across multiple antigen models, demonstrating a powerful method to boost immunogenicity for cancer vaccines or intracellular pathogens [34].
- For Protein-Replacement Therapies (Low Immunogenicity): To minimize unwanted immune activation and maximize protein expression, lipids can be designed for reduced reactogenicity. Furthermore, innovations in surface lipid chemistry are emerging. For instance, replacing the conventional PEG-lipid with a poly(2-ethyl-2-oxazoline) (POZ)-lipid has been shown in preclinical studies to prevent the induction of anti-PEG IgM and IgG antibodies upon repeated dosing, addressing a key safety concern related to anaphylaxis [35].
Essential Analytical and Experimental Methods
Characterizing the physicochemical properties and biological effects of novel ionizable lipids is essential for rational design. The following experimental protocols are standard in the field.
Key Experimental Protocols
Protocol 1: In Vitro Protein Expression Assay
- Objective: To quantify the translational efficiency of mRNA delivered by novel LNPs.
- Methodology:
- Cell Transfection: Transfect relevant cell lines (e.g., primary human skeletal muscle myoblasts - HSKM, or dendritic cells) with LNP formulations encapsulating reporter or antigen-encoding mRNA across a range of doses (e.g., 0.1-100 ng/well) [28].
- Incubation: Culture cells for 24-48 hours to allow for cellular uptake, endosomal escape, and protein translation.
- Detection: Quantify protein expression using flow cytometry (for surface or intracellular proteins), immunofluorescence imaging, or Western blot [28].
- Data Interpretation: Compare the level of protein expression against benchmark LNPs (e.g., SM-102 or MC3). m1Ψ-modified mRNA often shows higher expression than unmodified mRNA, but this is dependent on the ionizable lipid and cell type [28].
Protocol 2: Innate Immune Profiling In Vivo
- Objective: To evaluate the innate immune response and reactogenicity of novel LNP formulations.
- Methodology:
- Animal Immunization: Administer LNP (with or without mRNA) intramuscularly or intravenously to mice. Include control groups (e.g., PBS, empty LNP, benchmark LNP) [24].
- Serum Collection: Collect blood via retro-orbital or other routes at early timepoints (e.g., 6-8 hours post-injection) [24].
- Cytokine/Chemokine Quantification: Analyze serum using a bead-based multiplex assay (e.g., LEGENDplex) to measure key inflammatory mediators such as IL-6, IFN-γ, MCP-1, and TNF-α [24].
- Data Interpretation: Elevated levels of these cytokines indicate robust innate immune activation. A retrospective analysis can correlate early cytokine production with the magnitude of subsequent adaptive immune responses [24].
Protocol 3: Global Transcriptomic Analysis (RNA-Seq)
- Objective: To gain an unbiased, system-level view of the cellular response to LNP transfection.
- Methodology:
- Treatment and Harvest: Transfect cells (e.g., HSKM) with test and control LNPs. Harvest cells at multiple timepoints (e.g., 1, 4, 24 hours) [28].
- RNA Sequencing: Extract total RNA and perform high-throughput RNA sequencing.
- Bioinformatic Analysis: Perform differential gene expression analysis and Gene Set Variation Analysis (GSVA) using databases like MSigDB. Focus on gene ontology (GO) terms related to "antiviral response," "response to virus," "cytokine signaling," and "protein translation" [28].
- Data Interpretation: This reveals the breadth of immune activation. LNPs often induce a strong signature of antiviral/interferon-stimulated genes (e.g., OAS family, MX1, IFIT family). Differences in lipid composition can shift this transcriptional profile significantly [28].
The Scientist's Toolkit: Key Research Reagents
Table 2: Essential Reagents for LNP Innate Immunity Research
| Reagent / Solution | Critical Function | Technical Note |
|---|---|---|
| Ionizable Lipids (e.g., SM-102, ALC-0315) | Core functional component of LNPs for mRNA delivery and adjuvanticity [2]. | Source from GMP-grade suppliers (e.g., Croda/Avanti) for translational studies [35]. |
| N1-methylpseudouridine (m1Ψ) modified mRNA | Enhances stability, reduces innate immune recognition, and improves translation [2] [23]. | Compare with unmodified uridine mRNA to dissect immune contributions. |
| LEGENDplex Bead-Based Immunoassay | Multiplex quantification of key cytokines/chemokines (e.g., IL-6, MCP-1, IFN-γ) from small serum volumes [24]. | Enables high-throughput, sensitive profiling of innate immune responses in vivo. |
| Primary Human Cells (e.g., HSKM, hDCs) | In vitro models for assessing cell-type-specific delivery, protein expression, and immune activation [28]. | Provides human-relevant data prior to animal studies. |
| Quanti-Blue SEAP Assay | A sensitive, colorimetric assay for quantifying NF-κB/IRF activation downstream of PRRs like TLRs [34]. | Useful for screening the intrinsic immunostimulatory capacity of lipid components or TLR agonists. |
Ionizable lipids are the cornerstone of mRNA-LNP technology, masterfully engineered to perform the dual roles of a delivery tool and an immune adjuvant. Their rational design requires a deep understanding of the structure-activity relationships that govern pKa, delivery efficiency, and the resulting innate immune profile. The field is rapidly evolving beyond traditional screening methods. The integration of AI-driven lipid design [33] and the strategic incorporation of immune potentiators like TLR agonists [34] represent the forefront of next-generation LNP development. Furthermore, the emergence of novel lipids aimed at reducing anti-carrier immunogenicity [35] highlights the diversification of the platform for broader therapeutic applications beyond vaccinology. As research continues to unravel the intricate dialogue between ionizable lipids and the innate immune system, the design of LNPs will become increasingly precise, enabling safer and more effective mRNA therapeutics for a wide spectrum of diseases.
The efficacy of messenger RNA (mRNA) as a therapeutic platform is fundamentally linked to its ability to evade the body's innate immune system while achieving robust and sustained protein expression [26]. Exogenous mRNA introduced into the body is recognized by pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs) and retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), which perceive it as a foreign molecule, triggering potent immune responses that can severely limit therapeutic protein output [26]. Consequently, mRNA sequence engineering is not merely an exercise in maximizing protein yield but a critical endeavor to design stealth transcripts that can operate undetected. This in-depth technical guide examines three pillars of mRNA design—codon optimization, untranslated region (UTR) engineering, and 5' cap structure selection—framed within the context of modulating the innate immune response. Advances in these areas, including algorithmic sequence design and novel chemical modifications, are paving the way for more effective and safer mRNA therapeutics.
5' Cap Structures: The First Line of Defense
The 5' cap is a modified nucleotide structure that is essential for the stability, translatability, and immune evasion of mRNA [36]. It protects the mRNA from degradation by 5' to 3' exonucleases and facilitates binding with the eukaryotic initiation factor 4F (eIF4F) complex, which is crucial for the initiation of translation [37]. Critically, the specific structure of the cap plays a definitive role in how the innate immune system recognizes the mRNA molecule.
Table 1: Comparison of mRNA 5' Cap Structures
| Cap Type | Chemical Structure | Impact on Translation | Impact on Innate Immunogenicity | Key Recognition Factors |
|---|---|---|---|---|
| Cap 0 | m^7GpppN [36] | Good | High | Recognized as foreign by PRRs, triggering immune signaling [37]. |
| Cap 1 | m^7GpppNm [36] (2'-O-methylation of the first nucleotide) | Excellent | Low | Mimics mature self-RNA, evading immune detection [36] [37]. |
| Cap 2 | m^7GpppNmNm [36] (2'-O-methylation of first two nucleotides) | Enhanced | Very Low | Further enhances stability and reduces immunogenicity [36]. |
Two primary methods are employed for capping in vitro transcribed (IVT) mRNA:
- Post-Transcriptional Capping: A multi-enzymatic process where capping enzymes and methyltransferases are used to process uncapped IVT mRNA. This method can be labor-intensive and may result in variable capping efficiency [36].
- Co-Transcriptional Capping: This more advanced approach utilizes cap analogs (e.g., CleanCap technology) that are included directly in the IVT reaction. This method produces mRNA with a high proportion (up to 94%) of the desired Cap 1 structure, streamlining production and enhancing performance [36].
Figure 1: Impact of 5' Cap Structures on Innate Immune Recognition. Cap 0 is readily detected by the innate immune system, while Cap 1 and Cap 2 structures mimic endogenous RNA, enabling evasion.
UTR Engineering: Balancing Stability and Translational Efficiency
The untranslated regions (UTRs) flanking the coding sequence are critical regulatory elements that profoundly influence mRNA stability, subcellular localization, and translational efficiency, all of which can indirectly influence immunogenicity by modulating the intensity and duration of protein expression.
5' UTR: The 5' UTR is primarily responsible for the initiation of translation [36]. Its sequence and secondary structure are critical, as stable secondary structures (e.g., hairpins) can inhibit ribosome binding and scanning, thereby reducing protein yield [37]. To optimize this process, 5' UTRs from highly expressed human genes, such as alpha-globin or beta-globin, are commonly used in therapeutic mRNAs [36] [37]. Furthermore, incorporating internal ribosomal entry sites (IRES) can boost translation by recruiting ribosomes independently of the 5' cap [37].
3' UTR: The 3' UTR governs mRNA stability and half-life by modulating susceptibility to nucleases and interactions with RNA-binding proteins [36] [37]. Similar to the 5' UTR, sequences from globin genes are frequently employed to enhance transcript stability. The 3' UTR can also be engineered to avoid AU-rich elements (AREs), which are known to promote rapid mRNA decay and can activate innate immune pathways [38].
Table 2: UTR Design Parameters and Functional Impact
| Parameter | Considerations for Immune-Focused Design | Common Solutions/Sequences |
|---|---|---|
| 5' UTR Length | Shorter sequences may reduce complex secondary structures that hinder translation initiation [36]. | Typically 53-218 nucleotides [36]. |
| 5' UTR Sequence | Avoidance of unintended start codons (uAUGs) in upstream open reading frames (uORFs) that can create immunogenic peptides [38]. | Human alpha-globin or beta-globin UTRs [37]. |
| 3' UTR Stability Elements | Engineering to include stability elements and exclude AREs that trigger mRNA decay and inflammation [38]. | Human alpha-globin or beta-globin UTRs [37]. |
| Global Secondary Structure | Increased secondary structure stability correlates with extended mRNA half-life, reducing the need for high dosing that can overwhelm immune tolerance [39]. | Algorithmic optimization (e.g., LinearDesign) to find sequences with minimal free energy [39]. |
Codon Optimization: Beyond Mere Expression
Codon optimization involves replacing rare codons in the coding sequence (CDS) with synonymous codons that are more frequently used by the host cell, thereby matching the tRNA pool and enhancing the efficiency and accuracy of translation [37]. This not only boosts protein yield but can also prevent ribosomal stalling, which has been linked to the production of aberrant, potentially immunogenic peptides [26].
The challenge of mRNA design is the astronomically large sequence space. For example, the SARS-CoV-2 spike protein can be encoded by approximately 2.4 × 10^632 different mRNA sequences [39]. Traditional codon optimization focuses solely on codon usage frequency but often neglects RNA structural stability.
The LinearDesign Algorithm: This algorithm represents a significant advance by jointly optimizing codon usage (using the Codon Adaptation Index (CAI) as a metric) and mRNA structural stability (as measured by minimum free energy (MFE)) [39]. It formulates the search space as a deterministic finite-state automaton (DFA) and uses principles from computational linguistics (lattice parsing) to efficiently find the optimal sequence. Experimental data shows that vaccines designed with LinearDesign for COVID-19 and varicella-zoster virus (VZV) resulted in significantly improved mRNA half-life, protein expression, and antibody titers (up to 128-fold increase in mice) compared to traditional codon-optimized benchmarks [39].
Figure 2: Codon Lattice for mRNA Sequence Optimization. Each path represents a possible mRNA sequence. Algorithms like LinearDesign efficiently find the optimal balance between codon usage (green = optimal, red = rare) and global secondary structure.
Experimental Protocols for Validating mRNA Design
Protocol: Assessing mRNA ImmunogenicityIn Vitro
Objective: To measure the innate immune activation potential of engineered mRNA constructs by quantifying cytokine production and PRR signaling.
- Cell Seeding: Seed human peripheral blood mononuclear cells (PBMCs) or reporter cell lines (e.g., HEK-Blue hTLR cells) in 96-well plates.
- mRNA Transfection: Transfect cells with equimolar amounts of the test mRNA constructs (e.g., Cap 0 vs. Cap 1, or codon-optimized vs. LinearDesign-optimized) using a standard lipid nanoparticle (LNP) formulation or a transfection reagent.
- Cytokine Quantification: After 18-24 hours, collect cell culture supernatants. Measure the levels of key cytokines (e.g., IFN-α, IFN-β, TNF-α, IL-6) using Enzyme-Linked Immunosorbent Assay (ELISA) or multiplex bead-based immunoassays.
- PRR Signaling Assay: In reporter cell lines engineered with PRR-responsive promoters (e.g., IFN-β promoter) driving a secreted embryonic alkaline phosphatase (SEAP) or luciferase gene, measure the reporter activity in the supernatant or lysates as a direct readout of pathway activation.
- Data Analysis: Normalize cytokine levels or reporter activity to the total protein concentration. Compare immune activation across different mRNA designs to identify constructs with reduced immunogenicity.
Protocol: Evaluating mRNA Stability and Protein Expression
Objective: To determine the half-life and translational output of engineered mRNA.
- In Vitro Stability Assay: Incubate mRNA in a simulated physiological buffer (e.g., containing RNases) or cell lysate at 37°C. Aliquot the solution at various time points (e.g., 0, 1, 2, 4, 8 hours).
- RNA Integrity Analysis: Analyze the aliquots by denaturing agarose gel electrophoresis or capillary electrophoresis (e.g., Bioanalyzer) to quantify the remaining full-length mRNA. The half-life can be calculated from the decay curve.
- Cellular Translation Assay: Transfert cells (e.g., HEK293 or HeLa) with the mRNA constructs. For direct quantification, use mRNA encoding a reporter protein (e.g., GFP, luciferase) and measure fluorescence or luminescence at multiple time points post-transfection. Alternatively, for therapeutic proteins, use ELISA or Western blot to quantify protein production from the supernatant or cell lysates.
The Scientist's Toolkit: Essential Reagents for mRNA Engineering
Table 3: Key Research Reagent Solutions for mRNA Engineering
| Reagent / Tool | Function in mRNA Engineering |
|---|---|
| T7 RNA Polymerase | The primary enzyme for in vitro transcription (IVT) to synthesize mRNA from a DNA template [26]. |
| CleanCap Analog | A co-transcriptional capping reagent that enables high-yield production of Cap 1 mRNA, crucial for reducing immunogenicity [36]. |
| Pseudouridine-5'-TP (ΨTP) | A modified nucleotide triphosphate used in IVT to replace uridine. Incorporation of Ψ or N1-methylpseudouridine (m1Ψ) significantly decreases innate immune activation and enhances mRNA stability and translation [26] [37]. |
| Poly(A) Polymerase | Enzyme used for enzymatic addition of the poly(A) tail to the 3' end of IVT mRNA, a method to enhance stability [37]. |
| LinearDesign Software | An algorithmic tool that simultaneously optimizes mRNA codon usage and secondary structure for enhanced stability and protein expression [39]. |
| Lipid Nanoparticles (LNPs) | The leading delivery vehicle for mRNA in vivo, protecting it from degradation and facilitating cellular uptake and endosomal escape [26] [40]. |
1 Introduction The efficacy of messenger RNA (mRNA) vaccines and therapeutics hinges on robust and sustained protein expression, which is directly influenced by the innate immune response to exogenous RNA delivery. Conventional linear mRNA platforms, while revolutionary, face challenges of transient expression and inherent immunogenicity. Within the context of innate immune sensing, two novel platforms—self-amplifying RNA (saRNA) and circular RNA (circRNA)—have emerged as promising strategies to enhance the duration and level of antigen production. saRNA achieves this by encoding its own replication machinery, while circRNA leverages a covalently closed structure that confers nuclease resistance. This technical guide delineates the core principles, experimental workflows, and immune interactions of these platforms, providing a foundational resource for their rational development.
2 Platform Fundamentals and Innate Immune Context The innate immune system possesses a sophisticated array of pattern recognition receptors (PRRs) that detect exogenous RNA as a pathogen-associated molecular pattern, triggering type I interferon (IFN) responses that can paradoxically both stimulate adaptive immunity and inhibit protein translation. The design of saRNA and circRNA platforms must navigate this immunological landscape.
2.1 Self-Amplifying RNA (saRNA)
- Molecular Architecture: saRNA vectors are derived from the genomes of positive-sense single-stranded RNA viruses, such as alphaviruses (e.g., Semliki Forest Virus, Sindbis Virus). The key design feature is the replacement of viral structural protein genes with the gene encoding the therapeutic or vaccine antigen. The replicase complex (non-structural proteins, nsP1-4) is retained, enabling intracellular RNA amplification [41] [42].
- Mechanism of Sustained Expression: Upon delivery and cytoplasmic entry, the saRNA is initially translated to produce the viral replicase. This enzyme complex then synthesizes a negative-sense RNA strand, which serves as a template for generating multiple new positive-sense RNA strands. These include both the original replicon RNA for further amplification and subgenomic mRNAs that are efficiently translated into the target antigen. This cycle leads to a massive increase in the intracellular number of antigen-encoding RNA molecules, resulting in prolonged and heightened antigen expression compared to non-replicating mRNA [41].
- Innate Immune Interplay: The replication process generates double-stranded RNA (dsRNA) intermediates, which are potent ligands for PRRs like MDA5 and RIG-I, as well as protein kinase R (PKR). This can trigger a robust IFN-β response and inflammation. While this intrinsic adjuvanticity can be beneficial for vaccine applications, excessive innate immune activation can lead to premature shutdown of translation and increased reactogenicity, presenting a key development challenge [41] [9].
2.2 Circular RNA (circRNA)
- Molecular Architecture: circRNA is a covalently closed, continuous loop lacking 5' caps and 3' poly(A) tails. Therapeutic circRNA is primarily synthesized in vitro using permuted intron-exon (PIE) systems based on Group I or Group II self-splicing introns. Translation is initiated through an internal ribosome entry site (IRES) placed upstream of the antigen's open reading frame [41] [43].
- Mechanism of Sustained Expression: The closed-loop structure confers profound resistance to exonucleases, which are the primary cellular machinery for degrading linear mRNA. This dramatically increases the RNA's half-life. Consequently, a single circRNA molecule can engage with ribosomes repeatedly over an extended period, leading to sustained, albeit potentially lower at any single moment, antigen production [43] [42].
- Innate Immune Interplay: Engineered circRNAs exhibit reduced immunogenicity compared to linear mRNA. Their lack of free ends helps them evade certain RNA sensors, and their circular structure is not a typical PAMP. Furthermore, they can be produced with minimal dsRNA contaminants, a major contributor to IFN activation. This "immuno-silent" profile allows for persistent protein expression without triggering a strong antiviral state that would halt translation, making circRNA particularly attractive for protein-replacement therapies [41] [43].
Table 1: Comparative Analysis of saRNA and circRNA Platforms
| Feature | Self-Amplifying RNA (saRNA) | Circular RNA (circRNA) |
|---|---|---|
| Molecular Structure | Linear, 5' cap, 3' poly-A tail (on antigen transcript), ~9-12 kb | Covalently closed loop, no cap/no tail, IRES-dependent translation, ~1-3 kb |
| Primary Mechanism for Sustained Expression | Intracellular RNA amplification via viral replicase | Exonuclease resistance leading to extended RNA half-life |
| Expression Kinetics | High-level, prolonged expression (weeks) | Lower-level, durable expression (weeks) |
| Key Innate Immune Profile | Immunostimulatory; potent IFN-β induction via dsRNA intermediates | Reduced immunogenicity; "immuno-silent" profile |
| Major Technical Challenge | Risk of excessive inflammation; complex sequence design | Optimization of IRES efficiency; scalable production |
| Ideal Application Context | Prophylactic vaccines where strong immunity is needed quickly | Therapeutic proteins, long-term vaccination, repeated dosing |
3 Decoding the Innate Immune Response: Experimental Workflows A detailed understanding of the innate immune response to these platforms is gleaned from sophisticated in vivo and in vitro studies. Key methodologies are outlined below.
3.1 Single-Cell Transcriptomic Profiling of the Injection Site This workflow is critical for mapping the initial cellular and molecular events post-immunization.
3.1.1 Experimental Protocol
- Vaccination: Administer LNP-formulated saRNA, circRNA, or control (e.g., empty LNP, saline) via intramuscular injection to animal models (e.g., BALB/c mice) [11].
- Tissue Harvest: At multiple time points post-injection (e.g., 2, 16, 40 hours), resect the muscle tissue at the injection site and the draining lymph nodes (dLNs) [11].
- Single-Cell Suspension: Digest tissues mechanically and enzymatically to create single-cell suspensions.
- Library Preparation & Sequencing: Use platforms like the 10x Genomics Chromium system to barcode and prepare single-cell RNA sequencing (scRNA-seq) libraries from the suspensions. Sequence the libraries to generate transcriptome-wide data for tens of thousands of individual cells [11].
- Bioinformatic Analysis:
- Cell Type Identification: Cluster cells based on gene expression patterns and annotate cell types (e.g., fibroblasts, dendritic cells, monocytes) using known marker genes.
- Differential Expression: Identify differentially expressed genes (DEGs) and pathways (e.g., inflammatory response, IFN-stimulated genes) between treatment groups.
- Component Deconvolution: Compare samples receiving full mRNA-LNP to those receiving empty LNP to dissect the contribution of the RNA versus the delivery vehicle to the observed immune activation [11].
3.1.2 Key Workflow Visualization The diagram below summarizes the core experimental workflow for profiling injection site responses.
Diagram 1: scRNA-seq Workflow for Immune Profiling
3.2 Functional Validation of Type I IFN Signaling To confirm the mechanistic role of IFN, researchers employ IFNAR blockade studies.
3.2.1 Experimental Protocol
- Animal Model: Utilize wild-type (C57BL/6J) and IFNAR-/- mice [9].
- IFNAR Blockade: Administer an anti-IFNAR monoclonal antibody (e.g., 2.5 mg per dose) via intraperitoneal injection to wild-type mice 24 hours before and 24 hours after vaccination. IFNAR-/- mice serve as a genetic control [9].
- Vaccination and Sampling: Immunize all groups with the RNA platform of interest.
- Immune Readouts:
- Innate Phase: Measure serum cytokines (e.g., IL-6, MCP-1) and immunophenotype innate cells in dLNs (e.g., activation of dendritic cells) 8-24 hours post-vaccination [24] [9].
- Adaptive Phase: Quantify antigen-specific CD8+ T cells (via intracellular cytokine staining or tetramer staining) and antigen-specific antibody titers (via ELISA) 1-2 weeks post-booster [9].
4 Signaling Pathways in RNA Platform-Induced Immunity The innate immune sensing of saRNA and circRNA follows distinct molecular pathways, culminating in different adaptive immune outcomes. The schematic below illustrates these pathways for an immunostimulatory platform like saRNA.
Diagram 2: Innate Immune Sensing of Immunostimulatory RNA
5 The Scientist's Toolkit: Key Research Reagents Successful research into these platforms relies on a suite of specialized reagents and tools.
Table 2: Essential Research Reagents and Materials
| Reagent/Material | Function & Role in R&D | Specific Examples & Notes |
|---|---|---|
| Ionizable Lipids | Critical component of LNPs for encapsulating RNA and enabling endosomal escape; the specific structure can influence immunogenicity [11] [44]. | ALC-0315; SM-102; bespoke synthetic lipids. Key for in vivo delivery. |
| Nucleoside-Modified mRNA | Base modification (e.g., 1-methylpseudouridine, m1Ψ) reduces innate immune recognition of linear mRNA and saRNA, enhancing translation [24] [9]. | Standard for modern mRNA vaccines. Used to fine-tune immunogenicity. |
| RNase R Treatment | An exonuclease used in vitro to digest linear RNA and validate the successful circularization of circRNA preps [41]. | Quality control step for circRNA production. |
| Anti-IFNAR mAb | Monoclonal antibody for in vivo blockade of type I IFN signaling. Critical for mechanistic studies of immune activation [9]. | Clone: MAR1-5A3; administered intraperitoneally. |
| LegENDplex Bead Assays | Multiplex bead-based immunoassay for simultaneous quantification of multiple cytokines/chemokines from serum or tissue culture supernatant [24]. | Used to profile innate immune responses (e.g., IL-6, MCP-1, IFN-γ). |
| Group I/II Intron Kits | Enzymatic systems for high-efficiency in vitro production of circRNA via the PIE method [43]. | Core technology for scalable circRNA synthesis. |
6 Quantitative Data Summary Direct comparative studies provide quantitative insights into the performance of these novel platforms relative to each other and to conventional mRNA.
Table 3: Summary of Key Quantitative Findings from Preclinical Studies
| Parameter | Self-Amplifying RNA | Circular RNA | Conventional Linear mRNA | Experimental Context (Citation) |
|---|---|---|---|---|
| Relative Neutralizing Antibody Titer | Comparable to Circ-RNA | Comparable to SAM-RNA | Typically the reference level | SARS-CoV-2 RBD in mice [41] |
| Antigen-Specific T-cell Response | Induces response | Significantly higher (TH1-biased) | Lower than Circ-RNA | SARS-CoV-2 RBD in mice [41] |
| Stability at 4°C | Less stable | Stable for 4 weeks | Varies with modification | Formulated vaccine [41] |
| Induction of Key Cytokines (e.g., IL-6) | High (due to LNP + dsRNA) | Lower | High (primarily LNP-driven) | Mouse immunization model [24] [9] |
| Impact of IFNAR Blockade on Adaptive Immunity | Not directly tested | Not directly tested | Significantly enhances T-cell and antibody responses | LNP-mRNA vaccine in mice [9] |
7 Conclusion The advent of saRNA and circRNA platforms represents a significant evolution in nucleic acid therapeutics, offering sophisticated solutions to the challenge of sustained expression. saRNA acts as a self-driven amplifier, but its immunostimulatory nature requires careful modulation. In contrast, circRNA functions as a durable, steady-state generator of protein, benefiting from its innate immuno-stealth properties. The choice between these platforms is not merely technical but strategic, hinging on whether the therapeutic goal is to harness or evade the innate immune system. Future optimization will involve further engineering to fine-tune their interaction with host immunity, paving the way for more effective vaccines, protein-replacement therapies, and genetically encoded medicines.
Balancing Act: Troubleshooting Immunogenicity and Optimizing Efficacy
The type I interferon receptor (IFNAR) signaling pathway plays a pivotal yet complex role in shaping immune responses to mRNA vaccines. While essential for initiating innate immunity, recent evidence reveals that robust IFNAR activation can paradoxically attenuate subsequent adaptive immune responses. This whitepaper examines the dual nature of IFNAR signaling in the context of lipid nanoparticle (LNP)-delivered mRNA vaccines, synthesizing current research on the mechanistic basis for this dichotomy. We present quantitative data from key studies, detailed experimental methodologies for investigating IFNAR function, and visualization of critical signaling pathways. Understanding this delicate balance is crucial for optimizing next-generation mRNA vaccine platforms, particularly as researchers develop strategies to harness beneficial IFNAR signaling while minimizing its suppressive effects on adaptive immunity.
The discovery that IFNAR signaling serves as both an initiator and regulator of immune responses to LNP-mRNA vaccines represents a fundamental shift in our understanding of mRNA vaccinology. While type I interferons (IFN-α/β) have long been recognized as crucial mediators of antiviral defense, their role in vaccine-induced immunity is more nuanced. The encapsulation of nucleoside-modified mRNA in lipid nanoparticles has established the LNP-mRNA platform as a revolutionary vaccine technology, yet the immunological mechanisms underlying its efficacy remain incompletely understood [9]. Central to this puzzle is the interferon-alpha/beta receptor (IFNAR), a heterodimeric complex composed of IFNAR1 and IFNAR2 subunits that is ubiquitously expressed on nucleated cells [45] [46].
IFNAR activation triggers a sophisticated signaling cascade, primarily through the Janus kinase-signal transducer and activator of transcription (JAK-STAT) pathway, leading to the expression of hundreds of interferon-stimulated genes (ISGs) [47] [45]. This response creates an antiviral state in infected and neighboring cells, but also modulates the function of diverse immune cell populations. Recent investigations have revealed that the mRNA component of LNP-mRNA vaccines—rather than the LNP delivery vehicle itself—is essential for triggering a potent IFNAR-dependent innate immune response [9] [11]. Surprisingly, this activation can attenuate, rather than enhance, the development of adaptive immunity, creating a paradoxical scenario where the very pathway meant to bolster immune defense may ultimately limit vaccine efficacy.
This whitepaper examines the dual nature of IFNAR signaling in the context of mRNA vaccines, exploring the mechanistic basis for its contrasting effects on immune activation. Within the framework of innate immune responses to exogenous mRNA delivery, we synthesize recent findings on IFNAR's role as a double-edged sword, provide detailed experimental approaches for its study, and discuss implications for future vaccine design.
Molecular Mechanisms of IFNAR Signaling
Canonical and Non-Canonical Signaling Pathways
The IFNAR signaling network encompasses both canonical and non-canonical pathways that collectively regulate diverse cellular responses. The canonical JAK-STAT pathway begins when type I interferons (IFN-α/β) bind to the IFNAR1-IFNAR2 heterodimeric receptor complex, inducing conformational changes that activate the associated tyrosine kinases JAK1 and TYK2 [45] [48]. These kinases subsequently phosphorylate STAT1 and STAT2 proteins, which form a heterotrimeric complex with IRF9 known as ISGF3 (IFN-stimulated gene factor 3). ISGF3 translocates to the nucleus and binds to interferon-stimulated response elements (ISREs) in the promoters of hundreds of IFN-stimulated genes (ISGs) [47] [45].
Beyond this canonical pathway, type I IFNs can activate alternative signaling cascades. Non-canonical pathways include phosphorylation and dimerization of STAT3, STAT4, STAT5, and STAT6, as well as activation of Map kinases, PI3-kinase, and other signal transduction pathways [47] [45]. IFN-β has been shown to signal through IFNAR1 independently of IFNAR2 via a distinct non-canonical pathway [47]. This signaling complexity allows type I IFNs to regulate diverse cellular processes including antiviral defense, antigen presentation, cell survival, and differentiation.
Biological Consequences of IFNAR Activation
IFNAR signaling orchestrates a multifaceted immune response with profound implications for vaccine immunology. The induction of ISGs establishes a cellular antiviral state through proteins like protein kinase R (PKR) which inhibits cellular translation, and 2′5′ OAS/RNaseL which degrades RNA [47]. Beyond these cell-intrinsic effects, IFNAR activation modulates immune cell function by:
- Enhancing antigen presentation through increased MHC class I and II expression, particularly on dendritic cells [47] [45]
- Promoting dendritic cell maturation and upregulating co-stimulatory molecules (CD40, CD80, CD86) [47]
- Facilitating immune cell recruitment via induction of chemokines including CCL2, CCL3, CCL4, and CCL5 [47]
- Activating natural killer (NK) cells and enhancing their cytotoxicity and IFN-γ production [47]
- Recruiting inflammatory monocytes to sites of inflammation and infection [47]
These immunomodulatory effects position IFNAR at the interface between innate and adaptive immunity, influencing the quality and magnitude of subsequent T and B cell responses to vaccination.
The Dual Nature of IFNAR in Adaptive Immunity
Enhancing Effects: Priming the Immune System
IFNAR signaling establishes a favorable environment for adaptive immune activation through several mechanisms. In dendritic cells, IFN-I stimulation enhances MHC II expression and antigen presentation capacity while upregulating co-stimulatory molecules essential for T cell activation [47] [45]. Type I IFNs promote the differentiation of plasmacytoid DCs into myeloid-derived DCs with superior T cell stimulatory capacity [47]. Furthermore, IFNAR activation induces CCR7 expression on antigen-bearing DCs, facilitating their migration to draining lymph nodes where they interact with and activate naive T cells [45].
Table 1: IFNAR-Mediated Enhancement of Adaptive Immunity
| Mechanism | Key Effectors | Immunological Outcome |
|---|---|---|
| Antigen Presentation Enhancement | Increased MHC I/II, CD40, CD80/86 [47] [45] | Improved antigen display to T cells |
| Dendritic Cell Maturation | CCR7 upregulation, costimulatory molecule expression [45] | Enhanced migration to lymph nodes and T cell priming |
| Lymphocyte Recruitment | Induction of CCL3, CCL4, CCL5 [47] | Recruitment of NK cells and T cells to infection sites |
| Monocyte Activation | CCL2-mediated recruitment, differentiation to DCs [47] | Enhanced antigen presentation and inflammatory response |
| CD8+ T Cell Expansion | STAT3-Granzyme B pathway activation [45] | Increased cytotoxic T lymphocyte activity |
Attenuating Effects: Suppressing Adaptive Responses
Paradoxically, despite these activating functions, robust IFNAR signaling can suppress adaptive immune responses. Recent research on LNP-mRNA vaccines demonstrates that the mRNA component triggers IFNAR-dependent innate activation which attenuates subsequent adaptive immunity [9]. In murine models, LNP-mRNA vaccination induces rapid dendritic cell activation, monocyte recruitment to draining lymph nodes, and systemic cytokine responses—all dependent on IFNAR signaling. Importantly, transient inhibition of IFNAR signaling significantly enhances antigen-specific CD8+ T cell frequencies and antigen-specific antibody titers [9] [49].
The suppressive effects manifest through several mechanisms. Chronic IFNAR signaling can lead to T cell exhaustion through induction of inhibitory receptors like PD-1 [50]. Additionally, sustained ISG expression may directly inhibit protein translation, potentially reducing antigen production from mRNA vaccines [9]. The timing, duration, and magnitude of IFNAR activation appear critical in determining whether net enhancement or suppression occurs, with acute signaling being beneficial and prolonged signaling detrimental to adaptive immunity.
Table 2: IFNAR-Mediated Attenuation of Adaptive Immunity
| Mechanism | Key Effectors | Immunological Outcome |
|---|---|---|
| T Cell Exhaustion | PD-1 upregulation [50] | Reduced T cell functionality and proliferation |
| Translation Inhibition | PKR activation, eIF2α phosphorylation [9] [47] | Reduced antigen production from mRNA vaccines |
| Immune Regulation | Anti-inflammatory cytokine induction | Suppressed T cell activation and expansion |
| Cell-Intrinsic PD-1 | JAK/STAT-mediated PD-1 expression in melanoma cells [50] | Potential resistance to immune checkpoint blockade |
Experimental Evidence: Key Studies and Quantitative Data
Recent investigations have provided compelling evidence for IFNAR's dual role in mRNA vaccine immunity. A 2025 study demonstrated that the mRNA component of LNP-mRNA vaccines—rather than the LNP or encoded antigen—is essential for inducing potent IFNAR-dependent innate immune activation [9] [49]. Using mRNAs encoding different proteins and non-coding sequences in murine models, researchers established that this response attenuates adaptive immunity, as transient IFNAR blockade enhanced antigen-specific CD8+ T cells and antibody titers [9].
Complementary research published in Nature Communications (2024) revealed that injection site fibroblasts are highly enriched with delivered mRNA and express IFN-β specifically in response to the mRNA component [11]. The mRNA-LNP, but not LNP alone, induced migratory dendritic cells high in IFN-stimulated genes at injection sites and draining lymph nodes. Local IFN-β blocking significantly decreased mRNA vaccine-induced cellular immunity, highlighting the importance of spatial regulation in IFNAR signaling outcomes [11].
Table 3: Quantitative Findings from Key IFNAR Studies in mRNA Vaccination
| Study Reference | Experimental Model | Key Quantitative Findings |
|---|---|---|
| Frontiers in Immunology (2025) [9] | Murine model, LNP-mRNA vaccination | Transient IFNAR inhibition increased antigen-specific CD8+ T cell frequencies and antibody titers |
| Nature Communications (2024) [11] | Single-cell RNA-seq of vaccine site in mice | 2%-46% of cells at injection site were spike mRNA-positive, predominantly fibroblasts; IFN-β blocking reduced cellular immunity |
| Nature Communications (2024) [50] | Human and murine melanoma lines | IFN-α/β treatment induced PD-1 gene and protein expression via JAK/STAT signaling and chromatin remodeling |
Methodological Approaches: Investigating IFNAR Function
Experimental Models and Systems
Research on IFNAR signaling employs diverse experimental models, each offering distinct advantages. Murine models, particularly C57BL/6J and IFNAR-/- (IFNAR-deficient) strains, enable investigation of IFNAR-dependent mechanisms in vivo [9]. These systems allow for controlled vaccination studies with detailed analysis of immune responses in lymph nodes, spleen, and blood. Human and murine melanoma cell lines have proven valuable for examining cancer cell-intrinsic IFNAR signaling and PD-1 regulation [50]. For single-cell transcriptomic analysis, female BALB/c mice challenged with LNP or LNP-mRNA provide comprehensive profiles of injection site responses [11].
IFNAR Manipulation Techniques
Several technical approaches enable precise manipulation of IFNAR signaling:
- Genetic knockout models: IFNAR-/- mice completely lack functional type I interferon signaling [9]
- Antibody-mediated receptor blockade: Anti-IFNAR monoclonal antibodies (e.g., 2.5mg IP 24hr pre- and post-immunization) [9]
- Small molecule inhibitors: Deucravacitinib (TYK2 inhibitor) dissolved in DMSO and administered in PEG-300:Tween-80 vehicle [9]
- Conditional knockout systems: Cell type-specific deletion of IFNAR subunits to dissect cell-autonomous effects
Readouts and Analytical Methods
Comprehensive immune monitoring employs multiple analytical techniques:
- Flow cytometry: Characterization of immune cell populations, activation markers, and intracellular cytokines
- Single-cell RNA sequencing: Profiling of 83,094 single-cell transcriptomes from injection sites and 8,507 from draining lymph nodes [11]
- ELISpot assays: Quantification of antigen-specific T cells via IFN-γ production [11]
- Plaque reduction neutralization tests (PRNT): Measurement of neutralizing antibody titers [11]
- Chromatin immunoprecipitation (ChIP): Assessment of transcription factor binding to gene regulatory elements [50]
- ATAC-seq: Analysis of chromatin accessibility changes following IFNAR activation [50]
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents for Investigating IFNAR Signaling
| Reagent/Cell Line | Specific Example | Research Application | Key Function |
|---|---|---|---|
| IFNAR-Deficient Mice | IFNAR-/- (#032045, Jackson Laboratory) [9] | In vivo vaccine studies | Complete absence of type I IFN signaling |
| Anti-IFNAR Antibody | I-401-100 (Leinco Technologies) [9] | Receptor blockade experiments | Transient inhibition of IFNAR signaling |
| JAK/TYK2 Inhibitors | Deucravacitinib (MedKoo Biosciences) [9] | Signaling pathway dissection | Inhibition of downstream JAK-STAT signaling |
| Human Melanoma Lines | A2058, A375, G361, MeWo [50] | Cancer cell-intrinsic PD-1 studies | Model for IFNAR-regulated PD-1 expression |
| Murine Melanoma Lines | B16-F10, YUMM1.7, YUMMER1.7 [50] | Preclinical immunotherapy studies | Syngeneic models for tumor-immune interactions |
| LNP Formulations | Ionizable lipid (ALC-0315), Cholesterol, DSPC, DMG-PEG [9] | mRNA vaccine delivery | Efficient cytoplasmic mRNA delivery |
Implications for mRNA Vaccine Design and Therapeutic Intervention
The dual nature of IFNAR signaling presents both challenges and opportunities for therapeutic development. In mRNA vaccine design, strategies to modulate IFNAR activation include:
- Temporal control: Transient IFNAR inhibition during initial vaccination may enhance adaptive responses without compromising antiviral defense [9]
- mRNA engineering: Further optimization of nucleoside modifications and purification to fine-tune IFNAR activation [9] [11]
- LNP composition: Adjusting ionizable lipids and other components to control innate immune activation [9] [11]
- Dosing strategies: Optimizing prime-boost intervals to leverage beneficial IFNAR signaling while minimizing suppressive effects
In cancer immunotherapy, the discovery that IFNAR signaling induces melanoma cell-intrinsic PD-1 expression [50] suggests that IFNAR1 or JAK/STAT inhibition might disrupt responses to immune checkpoint blockade. This has important implications for patients receiving JAK inhibitors for autoimmune conditions who may have altered responses to cancer immunotherapies.
IFNAR signaling represents a paradigm of the delicate balance inherent in immune regulation—a double-edged sword that must be carefully managed in therapeutic contexts. In mRNA vaccination, the very same pathway that initiates essential innate immune activation can ultimately attenuate adaptive responses if unchecked. The molecular mechanisms underlying this dichotomy involve complex spatial and temporal regulation of JAK-STAT signaling, ISG expression, and subsequent effects on antigen presentation, lymphocyte function, and immune regulation. Future research should focus on developing precise strategies to harness the beneficial aspects of IFNAR signaling while mitigating its suppressive effects, potentially through engineered mRNA constructs, optimized delivery systems, or timed adjunct therapies. As mRNA vaccine platforms expand to address diverse infectious diseases and cancers, mastering the dual nature of IFNAR signaling will be essential for maximizing vaccine efficacy and advancing immunotherapeutic interventions.
Strategies to Overcome Inflammatory Reactogenicity and Individual Tolerability
The success of mRNA vaccine platforms, as demonstrated during the COVID-19 pandemic, represents a breakthrough in modern vaccinology. However, their widespread application is challenged by inflammatory reactogenicity—the physical manifestation of innate immune activation following vaccination [2] [51]. Reactogenicity encompasses local symptoms (pain, redness, swelling at the injection site) and systemic symptoms (fever, myalgia, headache) that, while typically self-limiting, can affect vaccine acceptance and individual tolerability [51]. These responses originate from the fundamental design of RNA vaccines: the mRNA molecules are recognized by innate immune sensors as potential pathogens, while the lipid nanoparticles (LNPs) used for delivery provide additional adjuvant activity [2] [11]. Balancing sufficient immunogenicity to achieve protective immunity while minimizing undesirable side effects represents a critical frontier in advancing mRNA vaccine technology. This whitepaper examines the mechanistic basis of reactogenicity and synthesizes current strategic approaches to overcome this challenge within the broader context of innate immune response to exogenous mRNA delivery.
Molecular Mechanisms of Innate Immune Recognition
Understanding reactogenicity requires examining the innate immune recognition pathways triggered by mRNA vaccine components. The response is initiated at the injection site, where both the LNP and the mRNA components activate distinct but complementary signaling cascades.
Sensing of mRNA and Lipid Nanoparticles
Intramuscularly administered mRNA-LNP vaccines are sensed by resident immune cells, stromal cells, and muscle cells at the injection site. Single-cell transcriptomic analyses reveal that the LNP component predominantly induces a pro-inflammatory response in stromal cells (fibroblasts, endothelial cells), characterized by the production of IL-6, TNF, and CCL2 [11]. Simultaneously, the mRNA component, even when nucleoside-modified, triggers type I interferon (IFN-β) responses, particularly in migratory dendritic cells (mDCs) [11]. This dual activation creates an inflammatory milieu that recruits monocytes, neutrophils, and other immune cells to the injection site, amplifying the local response and contributing to systemic symptoms when inflammatory mediators enter the circulation.
The sensing occurs through multiple pattern recognition receptors (PRRs). mRNA molecules can be detected by Toll-like receptors (TLR7/8) in endosomes, while double-stranded RNA (dsRNA) byproducts—inherent to in vitro transcription or formed during self-amplifying RNA (saRNA) replication—are recognized by TLR3, RIG-I, MDA5, and PKR in the cytosol [52] [2]. LNPs, particularly their ionizable lipid components, contribute significantly to adjuvanticity through mechanisms that may involve indirect activation of inflammasome pathways or other IL-1β activating pathways, though the precise receptors remain an active area of investigation [2].
Table 1: Innate Immune Sensors for mRNA Vaccine Components
| Vaccine Component | Sensing Mechanisms | Key Sensors | Downstream Effects |
|---|---|---|---|
| mRNA | 5' cap structure recognition | IFIT1, RIG-I | Translation inhibition, IFN production |
| Uridine recognition | TLR7/8 | Pro-inflammatory cytokine production | |
| dsRNA byproducts | TLR3, RIG-I, MDA5, PKR | Type I IFN, translational inhibition | |
| Lipid Nanoparticle (LNP) | Ionizable aminolipid | Proposed: inflammasome, other IL-1β pathways | IL-6, CCL2, other inflammatory cytokines |
Visualization of Innate Immune Signaling Pathways
The following diagram illustrates the key innate immune signaling pathways activated by mRNA-LNP vaccines:
Strategic Approaches to Mitigate Reactogenicity
Multiple innovative strategies are being developed to control reactogenicity while preserving or even enhancing immunogenicity. These approaches target different stages of the innate immune activation cascade, from vaccine component engineering to the incorporation of immunomodulators.
RNA Engineering and Formulation Optimization
The molecular design of RNA and delivery vehicles represents the first line of defense against excessive reactogenicity. Nucleoside modification (e.g., replacement of uridine with N1-methylpseudouridine, m1Ψ) remains a foundational strategy to reduce TLR7/8 recognition while enhancing translational capacity [2]. For self-amplifying RNA (saRNA) platforms, which generate dsRNA intermediates that potently activate innate sensing, the incorporation of 5-methylcytosine (5mC) has shown promise in partially reducing innate signaling without compromising replication [52]. Additionally, stringent purification to remove dsRNA contaminants from in vitro transcription reactions and the use of cap analogs that mimic natural eukaryotic cap structures help minimize unintended immune activation [2].
Delivery system optimization also plays a crucial role. While LNPs are essential for mRNA delivery, their reactogenicity profile can be modulated through ionizable lipid structure and LNP surface properties. Biodegradable lipids with ester linkages (e.g., SM-102, ALC-0315) have improved tolerability profiles while maintaining delivery efficiency [2]. Alternative delivery platforms, such as polysaccharide-based particles like Advax (derived from δ-inulin), show promise in providing adjuvant activity with reduced reactogenicity, as demonstrated in tuberculosis vaccine candidates [53].
Incorporation of Innate Immune Modulators
A particularly innovative approach involves the co-delivery of innate immune modulators that specifically counterbalance excessive inflammatory responses. Recent research has demonstrated the efficacy of encoding the Cardiovirus leader protein (dubbed "RNAx") from a discrete mRNA co-administered with the vaccine antigen [52]. This protein, which broadly dampens innate signaling by modulating nucleocytoplasmic transport (NCT), reduced interferon production and proinflammatory cytokines while enhancing antigen expression in both primary human cells and murine models.
When delivered in trans with an saRNA-LNP influenza vaccine, RNAx potently decreased serum biomarkers of reactogenicity while maintaining the magnitude of antibody and cellular responses. In some cases, it even enhanced binding antibody and neutralization titers post-boost, demonstrating that controlled innate modulation can improve both tolerability and immunogenicity [52]. This approach represents a paradigm shift from simply minimizing innate activation to actively shaping the quality of the immune response.
Table 2: Quantitative Outcomes of Reactogenicity Reduction Strategies
| Strategy | Experimental Model | Key Efficacy Metrics | Immunogenicity Impact |
|---|---|---|---|
| RNAx (Cardiovirus leader protein) | Mouse model with saRNA-LNP influenza vaccine | 170-fold enhancement in antigen expression; suppression of 14/15 saRNA-induced cytokines [52] | Maintained antibody and cellular responses; enhanced neutralization titers in some conditions [52] |
| Nucleoside modification (m1Ψ) | Human clinical trials (COVID-19 mRNA vaccines) | Reduced systemic reactogenicity compared to unmodified mRNA; maintained translation efficiency [2] | Robust neutralizing antibodies and T cell responses leading to high vaccine efficacy [2] |
| Advax adjuvant system | Mouse model of pulmonary tuberculosis | Reduced local reactogenicity vs. MPL/DDA adjuvant while maintaining protection [53] | Robust polyfunctional CD4+ T cell responses comparable to more reactogenic adjuvants [53] |
Experimental Approaches for Assessing Reactogenicity
In Vitro Screening Models
Initial assessment of reactogenicity potential employs primary human cell systems that recapitulate key aspects of innate immune sensing. Peripheral blood mononuclear cells (PBMCs) treated with candidate mRNA-LNP formulations provide a comprehensive platform for quantifying cytokine and interferon production using multiplex assays [52]. Specific cell lines, such as BJ human diploid fibroblasts (which possess intact innate sensing pathways), help evaluate the impact of interventions on gene of interest (GOI) expression in the context of innate signaling [52]. In these systems, the efficacy of innate immune modulators like RNAx can be quantified by measuring suppression of IFN-α, IFN-γ, IP-10, and other inflammatory mediators while monitoring antigen expression levels.
In Vivo Models and Biomarker Assessment
Animal models, typically mice, enable comprehensive evaluation of local and systemic reactogenicity. Key methodologies include:
- In vivo imaging to quantify antigen expression kinetics over time, comparing formulations with and without immunomodulatory components [52]
- Multiplex cytokine analysis of serum or plasma to quantify systemic inflammatory biomarkers
- Histopathological examination of injection sites to assess local inflammation and immune cell infiltration
- Single-cell RNA sequencing of injection site tissues to comprehensively map cellular responses and identify the specific contributions of LNP versus mRNA components [11]
The experimental workflow below outlines a comprehensive assessment approach:
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for Reactogenicity Research
| Reagent / Tool | Function/Application | Key Characteristics | Example Use Cases |
|---|---|---|---|
| N1-methylpseudouridine (m1Ψ) | Nucleoside modification | Reduces TLR7/8 recognition; enhances translation | Standard component of modern mRNA vaccines [2] |
| Ionizable lipids (SM-102, ALC-0315) | LNP component for mRNA delivery | Enable endosomal escape; biodegradable ester linkages | Pfizer/BioNTech and Moderna COVID-19 vaccines [2] |
| RNAx mRNA | Innate immune modulator | Encodes Cardiovirus leader protein; modulates NCT | Co-delivery with saRNA vaccines to suppress IFN while maintaining immunogenicity [52] |
| Advax adjuvant | Polysaccharide-based adjuvant | δ-inulin derived; enhances immunogenicity with low reactogenicity | Subunit vaccines (TB, hepatitis B) as alternative to more reactogenic adjuvants [53] |
| Cytokine multiplex arrays | Biomarker quantification | Simultaneous measurement of 40+ cytokines/chemokines | Comprehensive profiling of inflammatory responses in PBMCs or serum [52] |
The strategic landscape for overcoming inflammatory reactogenicity in mRNA vaccines is rapidly evolving from simple component optimization to sophisticated immune modulation. The most promising approaches recognize that innate immune activation exists on a spectrum—complete abolition may compromise immunogenicity, while excessive activation drives unacceptable reactogenicity. Future advances will likely involve personalized approaches that account for individual variation in innate immune sensing, potentially guided by biomarkers that predict tolerability profiles. Additionally, novel delivery systems with enhanced tissue specificity and reduced inflammatory properties, combined with fine-tuned immunomodulators that target specific innate pathways without broad immunosuppression, represent the next frontier. As our understanding of the intricate balance between immunogenicity and reactogenicity deepens, the development of more tolerable yet highly effective mRNA vaccines will expand their application across diverse populations and therapeutic areas.
The therapeutic efficacy of mRNA-based drugs is critically dependent on the yield and longevity of the translated protein product. mRNA translation boosters represent a novel class of adjuvant compounds that precisely modulate mRNA expression kinetics to enhance protein production. These boosters are classified as small-molecule compounds and macromolecular agents that improve translational fidelity through mechanisms including blockade of pattern recognition receptors, modulation of inflammatory cascades, facilitation of endosomal escape, and protection against enzymatic degradation [54]. The concept has gained significant traction following the clinical validation of COVID-19 mRNA vaccines, with these boosters now demonstrating expanded utility in gene editing therapies and protein replacement applications [54].
The development of mRNA translation boosters addresses a fundamental challenge in nucleic acid therapeutics: the delicate balance between achieving sufficient protein expression for therapeutic effect while managing the innate immune response to exogenous mRNA. When native mRNA is introduced into the human body, it triggers heterologous immune responses and undergoes rapid degradation, severely limiting its therapeutic applicability [26]. Translation boosters provide a strategic solution to this challenge by creating a more favorable intracellular environment for mRNA translation and persistence.
Innate Immune Recognition of Exogenous mRNA: The Central Challenge
The host immune system possesses an intricate network of Pattern Recognition Receptors (PRRs) evolved to detect conserved molecular patterns associated with pathogens. For mRNA vaccines and therapeutics, both the synthetic mRNA payload and components of the delivery system can engage these PRRs, triggering innate immune signaling that ultimately attenuates protein expression [9] [55].
Key RNA-Sensing Pathways
- Toll-like Receptors (TLRs): TLR3, located in endosomes, detects double-stranded RNA (dsRNA), while TLR7 and TLR8 recognize single-stranded RNA (ssRNA), particularly uridine-rich or GU-rich sequences [55]. Activation of these TLRs triggers downstream signaling that produces type I interferons and other pro-inflammatory cytokines.
- RIG-I-like Receptors (RLRs): Cytosolic receptors including RIG-I and MDA5 detect viral RNA and initiate signaling cascades that lead to type I interferon production [26] [55].
- Other Sensors: Additional sensors like protein kinase R (PKR) and 2'-5'-oligoadenylate synthetase (OAS) contribute to the antiviral state that inhibits mRNA translation [11].
The innate immune response creates a fundamental tension for mRNA therapeutics. While some immune activation may be beneficial for vaccine applications, excessive inflammation and interferon responses can significantly reduce protein yield by global inhibition of translation and increased mRNA degradation [9]. Recent studies have demonstrated that even with nucleoside-modified mRNA, the mRNA component itself—rather than just the LNP delivery vehicle—remains essential for triggering potent innate immune responses characterized by rapid activation of dendritic cells and recruitment of monocytes to draining lymph nodes [9].
Table 1: Key Innate Immune Sensors for Exogenous mRNA
| Receptor Class | Specific Receptors | Location | Ligand Specificity |
|---|---|---|---|
| Toll-like Receptors | TLR3, TLR7, TLR8 | Endosomal | dsRNA (TLR3), ssRNA (TLR7/8) |
| RIG-I-like Receptors | RIG-I, MDA5 | Cytosolic | Viral RNA structures |
| Kinase Systems | PKR | Cytosolic | dsRNA |
| Oligoadenylate Synthetases | OAS | Cytosolic | dsRNA |
Interferon-Mediated Translation Suppression
A critical mechanism by which innate immune responses limit protein yield is through type I interferon (IFN) signaling. Upon recognition of exogenous mRNA, PRR signaling activates transcription factors that induce IFN-β and other interferon-stimulated genes (ISGs) [11]. These interferons establish an antiviral state in cells that includes phosphorylation of eukaryotic initiation factor 2α (eIF2α), which globally inhibits translation initiation, and induction of RNA-degrading enzymes [9].
Recent single-cell transcriptome studies of mRNA vaccine injection sites have revealed that injection site fibroblasts are highly enriched with delivered mRNA and specifically express IFN-β in response to the mRNA component [11]. This IFN-β response induces migratory dendritic cells highly expressing interferon-stimulated genes (mDC_ISGs) at the injection site and draining lymph nodes. While this immune activation contributes to the adjuvant effect of mRNA vaccines, it simultaneously attenuates antigen production by creating a hostile environment for mRNA translation and persistence [9].
Mechanisms of mRNA Translation Boosters
Translation boosters employ diverse strategies to enhance protein yield by countering specific barriers in the mRNA delivery and expression pathway. These mechanisms can be categorized into several complementary approaches.
Innate Immune Modulation
The most prominent mechanism involves suppression of pattern recognition receptor signaling and subsequent interferon responses. Small molecules in this category include:
- PRR Antagonists: Compounds that directly inhibit TLR7/8, RIG-I, or other RNA sensors, preventing their activation by exogenous mRNA [54].
- Kinase Inhibitors: Molecules that target downstream signaling components such as TBK1, IKKε, or JAK/STAT pathways involved in interferon production and signaling [9].
- Interferon Receptor Blockers: Monoclonal antibodies or small molecules that temporarily inhibit IFNAR signaling [9].
Research has demonstrated that even brief and transient inhibition of IFNAR signaling significantly enhances the ability of LNP-mRNA vaccines to elicit adaptive immune responses, as evidenced by increased frequencies of antigen-specific CD8+ T cells and elevated titers of antigen-specific antibodies [9]. This approach allows the mRNA to bypass the interferon-mediated translation suppression while still leveraging other beneficial aspects of immune activation.
Endosomal Escape Enhancement
A critical bottleneck in mRNA delivery is the efficient release of mRNA from endosomes into the cytoplasm where translation occurs. Translation boosters can facilitate this process through:
- Endosomolytic Agents: Small molecules that promote endosomal membrane disruption through pH-dependent conformational changes or membrane-active properties [54].
- Ionizable Lipid Optimizers: Compounds that enhance the performance of ionizable lipids in LNPs, improving their endosomal escape efficiency [26].
These enhancers work synergistically with the LNP delivery system to increase the fraction of mRNA molecules that successfully reach the cytosol, thereby increasing the functional mRNA dose available for translation.
mRNA Stability and Translation Enhancement
Another approach focuses on directly enhancing mRNA stability and translational efficiency through:
- Nucleoside Analogues: Modified nucleotides such as pseudouridine (Ψ) and N1-methylpseudouridine (m1Ψ) that reduce immunogenicity while improving mRNA stability and translation efficiency [26]. These modifications significantly decrease recognition by innate immune sensors while maintaining or enhancing ribosomal engagement.
- Translation Initiation Promoters: Small molecules that enhance the activity of translation initiation factors or reduce the phosphorylation of eIF2α [54].
- Cap-Binding Complex Stabilizers: Compounds that enhance the interaction between the mRNA 5' cap and eukaryotic initiation factor 4E (eIF4E) [26].
Table 2: Categories of mRNA Translation Boosters and Their Mechanisms
| Booster Category | Representative Agents | Primary Mechanism | Effect on Protein Yield |
|---|---|---|---|
| PRR Antagonists | TLR7/8 inhibitors, RIG-I antagonists | Block innate immune recognition of mRNA | Reduces interferon-mediated translation suppression |
| Interferon Signaling Inhibitors | Anti-IFNAR antibodies, JAK inhibitors | Inhibit downstream IFN signaling | Prevents translation inhibition and mRNA degradation |
| Endosomal Escape Enhancers | Endosomolytic compounds, lipid optimizers | Facilitate mRNA release from endosomes | Increases cytosolic mRNA availability |
| Nucleoside Modifications | N1-methylpseudouridine, pseudouridine | Reduce immunogenicity, enhance stability | Improves mRNA half-life and translational efficiency |
| Translation Initiation Promoters | eIF4E stabilizers, eIF2α dephosphorylation promoters | Enhance translation initiation | Increases ribosome loading on mRNA |
Experimental Evaluation of Translation Boosters
In Vitro Assessment Protocols
Protocol 1: High-Throughput Screening of Translation Boosters
- Cell Seeding: Plate HEK-293 or HeLa cells in 96-well or 384-well plates at optimized densities (e.g., 10,000 cells/well for 96-well format) and culture for 24 hours.
- mRNA Transfection: Transfect cells with reporter mRNA (e.g., firefly luciferase, GFP) using LNP formulations or standard transfection reagents. Include appropriate controls (untreated, empty LNP, etc.).
- Booster Compound Application: Add candidate translation boosters at varying concentrations (typically 1 nM-10 μM) concurrently with or following mRNA transfection.
- Readout Measurement:
- Luminescence/Absorbance: For luciferase reporters, measure activity at 6, 24, 48, and 72 hours post-transfection using appropriate substrates.
- Flow Cytometry: For fluorescent protein reporters, analyze the percentage of positive cells and mean fluorescence intensity at various time points.
- ELISA: For specific antigens, quantify protein production using standardized ELISA protocols.
- Viability Assessment: Perform parallel MTT, CCK-8, or ATP-based assays to exclude cytotoxic effects.
- Immune Marker Analysis: Quantify interferon and cytokine production using multiplex ELISA or RNA quantification of ISGs to correlate protein yield with immune modulation [11] [9].
Protocol 2: Mechanism of Action Studies
IFNAR Blocking Experiments:
- Pre-treat cells with anti-IFNAR monoclonal antibodies (2.5 mg/mL) 24 hours prior to mRNA transfection [9].
- Transfert with reporter mRNA in the presence or absence of candidate boosters.
- Compare protein yield with and without IFNAR blockade to determine interferon dependence.
Endosomal Escape Quantification:
- Use fluorescence-based assays with labeled mRNA to track cellular localization.
- Employ Gal8- or Gal3-based assays to monitor endosomal disruption.
- Compare endosomal escape efficiency with and without booster compounds.
Ribosome Profiling:
- Perform Ribo-seq analysis to assess ribosome occupancy and translation elongation rates [56].
- Identify potential ribosomal stalling or frameshifting issues induced by booster compounds.
In Vivo Evaluation Methods
Protocol 3: Murine Model Assessment of Translation Boosters
- Animal Models: Use 6-8 week old female C57BL/6J or BALB/c mice, age-matched between experimental groups [11] [9].
- Vaccination and Booster Administration:
- Formulate mRNA-LNP vaccines with or without candidate booster compounds.
- Administer via intramuscular injection (50 μL per hind leg) [9].
- For systemic delivery, use intravenous injection via tail vein.
- Tissue Collection and Analysis:
- Euthanize animals at predetermined time points (2, 6, 16, 24, 40 hours post-injection) [11].
- Collect injection site tissues, draining lymph nodes, and serum samples.
- Process tissues for single-cell RNA sequencing, immunohistochemistry, or protein analysis.
- Immune Monitoring:
- Protein Expression Quantification:
- Measure antigen production in tissues using Western blot, ELISA, or immunohistochemistry.
- Compare expression kinetics and duration between booster-treated and control groups.
Signaling Pathways in mRNA Translation and Immune Recognition
The efficacy of mRNA translation boosters can be understood through their modulation of key signaling pathways involved in mRNA translation and immune recognition.
Diagram 1: Signaling Pathways in mRNA Translation and Immune Recognition. Translation boosters (green) enhance protein yield by modulating key steps in immune recognition and protein synthesis pathways.
Advanced Booster Strategies and Future Directions
Computational Optimization Approaches
Recent advances in computational methods have enabled more sophisticated approaches to enhance mRNA translation. RiboDecode represents a deep learning framework that generates optimized mRNA codon sequences for enhanced translation by directly learning from large-scale ribosome profiling data [56]. This approach explores a vast sequence space beyond the limitations of traditional rule-based optimization methods like codon adaptation index (CAI).
Key innovations in computational optimization include:
- Context-Aware Optimization: Algorithms that consider cellular environment, including expression of RNA-binding proteins and translation factors [56].
- Joint Optimization of Multiple Parameters: Simultaneous optimization of translation efficiency and mRNA stability through minimum free energy (MFE) predictions [56].
- Generative Sequence Exploration: Deep learning models that explore novel codon sequences beyond human-designed patterns [56].
In vitro experiments with computationally optimized sequences have shown substantial improvements in protein expression, significantly outperforming previous methods. In vivo studies demonstrated that optimized influenza hemagglutinin mRNAs induced approximately ten times stronger neutralizing antibody responses compared to unoptimized sequences [56].
Novel mRNA Formats and Their Implications
Beyond conventional linear mRNA, novel mRNA structures present both opportunities and challenges for translation boosting:
- Self-Amplifying RNA (saRNA): Engineered from alphavirus genomes, saRNA replicates itself within cells, enabling prolonged protein expression and potentially reducing dose requirements [26].
- Circular RNA (circRNA): Lacking free ends, circRNA evades exonuclease-mediated degradation and shows extended half-life, making it particularly suitable for applications requiring sustained protein production [26].
- Multitailed mRNA: Novel structural configurations that may enhance stability and translational efficiency through unknown mechanisms [26].
Each of these formats interacts differently with the innate immune system and translation machinery, potentially requiring specialized booster compounds tailored to their unique properties.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for mRNA Translation Booster Studies
| Reagent Category | Specific Examples | Research Application | Key Function |
|---|---|---|---|
| Nucleoside-Modified mRNAs | N1-methylpseudouridine (m1Ψ), Pseudouridine (Ψ) | mRNA formulation | Reduce immunogenicity, enhance translation efficiency |
| Lipid Nanoparticles | ALC-0315, DLin-MC3-DMA, SM-102 | mRNA delivery | Protect mRNA, facilitate cellular uptake and endosomal escape |
| PRR Inhibitors | TLR7/8 antagonists (e.g., IRS954), RIG-I inhibitors | Mechanism studies | Block specific innate immune recognition pathways |
| Interferon Signaling Modulators | Anti-IFNAR antibodies, JAK inhibitors (e.g., Deucravacitinib) | Functional validation | Temporarily inhibit IFN signaling to enhance translation |
| Reporter Systems | Firefly luciferase, GFP, nanoluciferase | Quantification of protein yield | Enable sensitive measurement of translation efficiency |
| Ribosome Profiling Kits | Ribo-seq library preparation kits | Translation mechanism studies | Map ribosome positions and quantify translation elongation |
| Single-Cell RNA Seq Kits | 10x Genomics Chromium, Parse Biosciences | Immune profiling | Characterize heterogeneous cellular responses to mRNA delivery |
mRNA translation boosters represent a transformative approach to enhancing the therapeutic efficacy of mRNA-based medicines by strategically modulating the complex interplay between protein expression and innate immune recognition. Through mechanisms including innate immune modulation, endosomal escape enhancement, and direct translation potentiation, these boosters address fundamental limitations in current mRNA technology.
The future of translation boosters lies in the development of context-specific compounds tailored to particular therapeutic applications—vaccines requiring balanced immune activation, protein replacement therapies needing maximal sustained expression, and gene editing applications demanding precise temporal control. As computational optimization methods advance and novel mRNA formats emerge, translation boosters will play an increasingly important role in unlocking the full potential of mRNA therapeutics across diverse medical applications.
The integration of sophisticated booster compounds with optimized mRNA sequences and advanced delivery systems promises to usher in a new generation of mRNA medicines with enhanced efficacy, reduced dosing requirements, and expanded therapeutic indications.
The convergence of artificial intelligence (AI) and nanomedicine has fundamentally transformed the development of lipid nanoparticles (LNPs) for mRNA delivery, enabling the precise engineering of formulations that navigate the complex landscape of innate immune recognition. Ionizable lipids serve as the pivotal component of LNPs, the leading non-viral messenger RNA delivery technology [57]. Traditional methods relying on experimental screening and rational design are being superseded by AI-driven approaches that can rapidly identify optimal structures from a vast chemical space. Furthermore, the innate immune system's response to exogenous mRNA presents both a challenge and an opportunity: uncontrolled activation can attenuate adaptive immunity, while appropriately modulated responses are essential for vaccine efficacy [9]. This technical guide explores the integration of in silico profiling and synthetic transcriptomics within a framework that acknowledges the critical role of innate immune signaling in LNP-mRNA vaccine performance. By adopting these advanced computational strategies, researchers can accelerate the development of next-generation genetic medicines with enhanced translational potential.
AI-Driven Methodologies for LNP Optimization
Machine Learning Frameworks for Virtual Lipid Screening
Machine learning (ML) algorithms have disrupted traditional nanomedicine workflows by enabling predictive modeling of multi-parametric interactions that govern LNP behavior. These approaches can extrapolate to structures divergent from their training sets, allowing for the discovery of novel lipid architectures with programmable properties [57]. The foundational requirement for implementing these strategies is a high-quality, extensive dataset of LNP activity measurements—preferably exceeding 15,000 lipid structures—to ensure model accuracy and generalizability [58].
Directed Message-Passing Neural Networks (D-MPNNs) represent a particularly powerful approach for lipid design. These networks operate directly on graph representations of lipid molecules, learning structure-property relationships by passing messages between atoms along chemical bonds. When trained on comprehensive datasets of LNP activity measurements (>9,000 data points), D-MPNNs can predict nucleic acid delivery efficiency both in vitro and in vivo with high accuracy [57]. The model processes each lipid structure through a series of neural network layers that capture increasingly complex molecular features, ultimately generating predictions of key performance metrics such as encapsulation efficiency, endosomal escape capability, and delivery efficacy.
Random Forest models offer an alternative approach, particularly effective for predicting critical physicochemical parameters like pKa values of ionizable lipids. These ensemble methods construct multiple decision trees during training and output the mean prediction of the individual trees, demonstrating strong performance with mean absolute error (MAE) values indicating high predictive accuracy [58]. The implementation typically involves:
- Feature Engineering: Calculation of molecular descriptors (e.g., molecular weight, logP, polar surface area) and fingerprint vectors that numerically encode lipid structures.
- Model Training: Optimization of tree parameters through cross-validation to prevent overfitting.
- Validation: External validation using held-out test sets to ensure generalizability to novel lipid structures.
Generative Adversarial Networks (GANs) expand the design space beyond virtual screening by creating entirely novel ionizable lipid structures with programmed characteristics. The GAN framework consists of two competing neural networks: a generator that produces candidate lipid structures and a discriminator that evaluates their authenticity compared to known effective lipids. Through iterative training, the generator learns to produce increasingly realistic and effective lipid designs, with demonstrated capability to generate 92% novel ionizable lipids with programmable pKa (6.2–6.8) and specific branching patterns [58]. Quantum computing-enhanced GANs represent the next frontier, potentially enabling the exploration of exponentially larger chemical spaces for lipid discovery.
Table 1: Performance Metrics of AI Models for LNP Design
| AI Model | Primary Application | Key Performance Metrics | Limitations |
|---|---|---|---|
| Directed Message-Passing Neural Networks | Structure-activity prediction for nucleic acid delivery | Accurate in vitro/in vivo delivery prediction; Identified FO-32/FO-35 lipids with superior lung delivery [57] | Requires large training datasets (>9,000 measurements) |
| Random Forest Models | pKa prediction and virtual screening | MAE < 0.65 for pKa prediction; R² > 0.85 for structure-property relationships [58] | Limited extrapolation beyond chemical space of training data |
| Generative Adversarial Networks (GANs) | De novo lipid design | 92% novel ionizable lipids with programmable pKa (6.2-6.8) [58] | Computational intensity; Challenge in objective function definition |
Algorithmic mRNA Design for Stability and Regulated Immunogenicity
The mRNA component itself represents a critical design factor that influences both stability and innate immune recognition. The LinearDesign algorithm addresses the prohibitive computational challenge of mRNA sequence optimization by adapting lattice parsing techniques from computational linguistics [39]. This approach formulates the mRNA design space using a deterministic finite-state automaton (DFA) that compactly encodes exponentially many mRNA candidates, with each path representing a possible sequence encoding the target protein.
The algorithm employs a lattice parsing approach to identify optimal mRNA sequences based on two primary objectives:
- Structural Stability Optimization: Finding the mRNA sequence with the lowest minimum-free-energy (MFE) change among all possible sequences encoding the target protein, using standard RNA folding energy models.
- Joint Stability and Codon Optimization: Balancing structural stability with codon optimality through the objective function: MFE – λ|p| log CAI, where λ is the CAI weight parameter and |p| is the protein length.
For the SARS-CoV-2 spike protein (1,273 amino acids), LinearDesign can identify optimal mRNA sequences in just 11 minutes, compared to the 10⁶¹⁶ billion years that enumeration would require [39]. Experimental validation demonstrates that vaccines designed using this approach substantially improve chemical stability in vitro, protein expression in cells, and immunogenicity in vivo, with COVID-19 vaccines achieving up to 128× the antibody response of codon-optimized benchmarks in murine models [39].
Experimental Profiling and Validation Frameworks
In Vitro and In Vivo Profiling Protocols
LNP Formulation and Characterization Protocol:
- Lipid Mixture Preparation: Combine ionizable lipid, cholesterol, distearoylphosphatidylcholine (DSPC), and DMG-PEG2000 at molar ratios (e.g., 40:47.5:10.5:2) in absolute ethanol [9].
- Aqueous Phase Preparation: Suspend mRNA payloads in citrate buffer (50mM, pH 4.5).
- Nanoparticle Assembly: Utilize microfluidic mixing systems (e.g., NanoAssemblr) with total flow rates of 12mL/min to ensure reproducible LNP formation.
- Dialysis and Buffer Exchange: Dialyze against PBS overnight to remove residual ethanol and achieve buffer exchange.
- Characterization Metrics:
- Size and Polydispersity: Dynamic light scattering (DLS) measurements; target hydrodynamic size of 60-70nm with PDI <0.2 [9].
- Encapsulation Efficiency: RiboGreen assay; target >93% encapsulation [9].
- Surface Charge: Zeta potential measurements; typically -8 to -9mV for stable formulations [9].
- mRNA Integrity: Electrophoretic or HPLC analysis to confirm structural integrity post-encapsulation.
In Vitro Transfection Efficiency Protocol:
- Cell Line Selection: Utilize relevant cell lines (e.g., HEK293, HeLa, or primary cells depending on target tissue).
- Dosing Strategy: Apply LNPs at varying mRNA concentrations (e.g., 0.1-1μg/mL) in serum-free or reduced-serum media.
- Incubation Period: 4-6 hours followed by replacement with complete growth media.
- Expression Analysis:
- Time Course Assessment: Measure protein expression at 24, 48, and 72 hours post-transfection.
- Quantitative Methods: Flow cytometry for fluorescent reporters, luciferase assays for enzymatic reporters, or ELISA for specific proteins.
- Cell Viability Assessment: MTT or CellTiter-Glo assays to evaluate cytotoxicity.
In Vivo Biodistribution and Efficacy Studies:
- Animal Models: Utilize murine models (e.g., C57BL/6J, BALB/c) or higher species (e.g., ferrets) for pulmonary delivery validation [57].
- Dosing Administration:
- Tissue Collection and Analysis:
- Time Points: Collect tissues (e.g., lung, liver, spleen, lymph nodes) at 6, 24, and 48 hours post-administration.
- mRNA Quantification: RT-qPCR analysis of target mRNA in tissue homogenates.
- Protein Expression: Immunohistochemistry or Western blot of tissue sections.
- Immune Response Characterization:
- Cytokine Profiling: Multiplex ELISA of serum or tissue homogenates for IFN-α, IFN-β, IL-6, TNF-α.
- Immune Cell Recruitment: Flow cytometry of draining lymph nodes for dendritic cell activation and monocyte recruitment.
Table 2: Key Research Reagents and Experimental Systems
| Category | Specific Reagents/Systems | Function/Application | Technical Notes |
|---|---|---|---|
| Ionizable Lipids | ALC-0315, SM-102, AI-designed FO-32/FO-35 [57] [9] | Core LNP component for mRNA encapsulation and endosomal release | pKa range 6.2-6.8 optimal for endosomal escape |
| Structural Lipids | Cholesterol, DSPC [9] | LNP stability and bilayer structure | 10-15 mol% typical for DSPC |
| PEGylated Lipids | DMG-PEG2000 [9] | LNP stability, reduce opsonization | 1.5-3 mol%; higher percentages reduce efficacy |
| mRNA Constructs | Nucleoside-modified (m1Ψ), cellulose-purified [9] [39] | Antigen expression with reduced immunogenicity | Remove dsRNA contaminants to minimize IFN response |
| Characterization Tools | NanoAssemblr, DLS, Zetasizer, RiboGreen [9] | LNP fabrication and QC | Target size 60-80nm, PDI <0.2, EE >90% |
| Cell-Based Systems | Precision-cut human lymph node slices [59] | Ex vivo human immune response modeling | Retains tissue architecture and native cell populations |
| Animal Models | C57BL/6J, IFNAR-/- mice, ferrets [57] [9] | In vivo efficacy and biodistribution | Ferrets model human-like lung physiology for pulmonary delivery |
Profiling Innate Immune Responses to LNP-mRNA Formulations
Understanding the innate immune recognition of LNP-mRNA formulations is essential for optimizing their efficacy and safety profiles. Critical experimental approaches include:
Type I Interferon Response Characterization:
- IFNAR Signaling Studies: Utilize IFNAR-/- mice to delineate interferon-dependent effects [9].
- Blocking Experiments: Administer anti-IFNAR monoclonal antibodies (2.5mg, i.p.) 24 hours pre- and post-immunization to transiently inhibit signaling [9].
- Downstream Signaling Analysis: Assess phosphorylation of STAT1/STAT2 and expression of interferon-stimulated genes (ISGs) via Western blot and RT-qPCR.
Draining Lymph Node (dLN) Analysis:
- Fine Needle Aspiration (FNA): Serial sampling of dLNs for cellular composition and activation status [59].
- Flow Cytometry Panel:
- Dendritic cell activation markers: CD80, CD86, MHC-II
- Monocyte recruitment: CD115, Ly6C
- Innate lymphoid cells: NK1.1, CD127
- Cytokine Milieu: Multiplex analysis of dLN homogenates for IL-1β, IFN-γ, CXCL8.
Precision-Cut Human Lymph Node Model:
- Tissue Acquisition: Obtain healthy human LNs from surgical procedures.
- Slice Preparation: Generate 300μm thick sections using precision tissue slicers.
- Ex Vivo Culture: Maintain slices in complete RPMI with LNP-mRNA stimulation.
- Single-Cell RNA Sequencing: Profile transcriptional responses across cell populations (T cells, B cells, monocytes, macrophages, stromal cells) [59].
- Multiplexed Imaging: Spatial analysis of immune cell interactions and cytokine production.
Integrating Immune Response Data into AI Models
Multi-Objective Optimization Framework
The integration of immune response parameters transforms LNP design from a单纯的 delivery efficiency optimization to a sophisticated balancing of multiple biological objectives. The comprehensive dataset must include:
- Delivery Efficiency Metrics: mRNA encapsulation efficiency, cellular uptake, protein expression levels.
- Innate Immune Parameters: Type I IFN induction, proinflammatory cytokine profiles, dendritic cell maturation markers.
- Adaptive Immune Outcomes: Antigen-specific antibody titers, CD8+ T cell frequencies, memory cell formation.
- Safety Indices: Local reactogenicity, systemic inflammatory markers, liver enzyme elevations.
SHapley Additive exPlanations (SHAP) values provide critical model interpretability, quantifying the contribution of each molecular feature to predicted immune responses. Models achieving SHAP values >0.65 demonstrate sufficient transparency for regulatory consideration and mechanistic insight [58].
Experimental Validation of AI-Designed Formulations
The AI-identified lipids FO-32 and FO-35 exemplify the success of this integrated approach. Experimental validation demonstrated:
- Local mRNA Delivery: Efficient delivery to mouse muscle and nasal mucosa [57].
- Pulmonary Efficacy: FO-32 matched state-of-the-art performance for nebulized mRNA delivery to mouse lung and efficiently delivered mRNA to ferret lungs [57].
- Immune Profile: Favorable innate immune activation patterns compatible with robust adaptive immunity.
The implementation of blockchain-enabled regulatory frameworks can integrate SHAP values for AI model interpretability with immutable audit trails of formulation parameters, ensuring compliance with evolving Good Manufacturing Practice (GMP) standards throughout the production lifecycle [58].
The integration of AI-guided LNP design with sophisticated immune profiling represents a paradigm shift in the development of mRNA therapeutics and vaccines. By simultaneously optimizing for delivery efficiency and controlled immunogenicity, researchers can overcome the historical limitations of the LNP-mRNA platform. The methodologies outlined in this technical guide provide a framework for the design and characterization of next-generation formulations that balance efficacy with favorable immune compatibility. As these technologies mature, the incorporation of quantum machine learning for stability prediction and edge computing for real-time formulation modifications will further accelerate the development of precision genetic medicines [58]. This integrated approach ultimately enables the exploration of previously unreachable design spaces, yielding LNPs with transformative potential for treating a broad spectrum of genetic, infectious, and neoplastic diseases.
The clinical success of mRNA vaccines during the COVID-19 pandemic has fundamentally transformed vaccinology, revealing both the profound potential and significant challenges of achieving protective immunity across diverse populations. Personalized vaccinology represents a paradigm shift from the traditional "one-size-fits-all" approach to immunization, instead focusing on the adjustment of vaccine strategies to account for individual variations in immune response. This approach is critically needed because multiple studies have demonstrated substantial interindividual variability in vaccine-induced immunity influenced by factors including age, genetic background, immunological history, and environmental exposures [60].
The foundation of personalized vaccinology rests upon understanding how innate immune recognition of vaccine platforms, particularly exogenous mRNA delivery, shapes subsequent adaptive immunity. The innate immune system serves as the body's first line of defense, relying on germline-encoded pattern recognition receptors (PRRs) to detect molecular signatures known as pathogen-associated molecular patterns (PAMPs) [21]. When mRNA vaccines are administered, their components are recognized by various PRRs, initiating a cascade of signaling events that ultimately determine the quality, magnitude, and durability of the antigen-specific adaptive immune response [2] [21]. This innate-adaptive immune interface provides critical opportunities for strategic intervention to overcome response heterogeneity and optimize vaccine efficacy for each individual.
Technical Foundations: mRNA Vaccine Immunology
Innate Immune Sensing of mRNA Vaccines
The lipid nanoparticle (LNP)-encapsulated, nucleoside-modified mRNA platform activates the innate immune system through multiple complementary mechanisms involving both the mRNA molecule itself and its delivery vehicle. The LNP carrier acts as a powerful adjuvant, inducing robust cytokine and chemokine responses that recruit and activate antigen-presenting cells [2]. Meanwhile, the mRNA component can be sensed by various intracellular receptors, though nucleoside modification (e.g., replacement of uridine with N1-methylpseudouridine (m1Ψ)) significantly reduces this recognition while enhancing translational capacity [2] [61].
Table 1: Innate Immune Sensing Mechanisms of mRNA Vaccine Components
| Vaccine Component | Sensing Mechanisms | Key Sensors | Resulting Immune Activation |
|---|---|---|---|
| mRNA | 5' cap structure recognition | IFIT1, RIG-I (for Cap0) | Translation inhibition, IFN production |
| Uridine-containing RNA | TLR7/8 | Proinflammatory cytokines, type I IFN | |
| dsRNA byproducts | TLR3, RIG-I, MDA5, PKR | Type I/III IFN, translational shutdown | |
| Lipid Nanoparticle (LNP) | Ionizable aminolipid | Inflammasome, unknown PRRs | IL-1β, IL-6, chemokines (CCL2, CCL3) |
| PEG-lipid | Possible complement activation | Local inflammation, immune cell recruitment |
Recent single-cell transcriptomic studies have revealed the complex cellular dynamics at mRNA vaccine injection sites, demonstrating that fibroblasts are highly enriched with delivered mRNA and serve as significant producers of IFN-β specifically in response to the mRNA component [11]. This IFN-β production creates a localized environment that promotes the activation of migratory dendritic cells highly expressing interferon-stimulated genes (mDC_ISGs), which are crucial for initiating adaptive immunity [11]. The coordinated interplay between stromal and immune cells at the injection site ultimately determines the strength and quality of the vaccine response.
From Innate Sensing to Adaptive Immunity
The bridge between innate immune activation and adaptive immunity represents the critical juncture where personalized strategies can exert their greatest influence. Upon sensing mRNA vaccine components, activated antigen-presenting cells (APCs) upregulate costimulatory molecules and migrate to draining lymph nodes, where they present processed antigens to naïve T cells [21]. The cytokine milieu established during innate sensing directs the differentiation of T helper cell subsets, thereby influencing the character of the antibody response and the generation of memory cells.
The paradigm of innate regulation of adaptive immunity is well-established, with antigen-specific immune responses being enhanced through several strategies: (1) inducing functional immune responses, (2) accelerating initial immune processes, (3) regulating the scope, specificity, or affinity of immune responses, and (4) enhancing immune memory effects [21]. Each of these strategies offers potential intervention points for addressing immune heterogeneity.
Current Research: Insights into Response Variability and Personalization Approaches
Heterologous Vaccination Strategies
The administration of different vaccine types in sequence (heterologous prime-boost) has emerged as a powerful strategy to enhance immune responses beyond what can be achieved with homologous regimens. Research comparing mRNA prime/protein boost versus protein prime/mRNA boost vaccination against influenza hemagglutinin has demonstrated that the sequence of vaccination critically directs immune responses [62]. Specifically, mRNA priming followed by protein boosting (R-P regimen) elicited balanced IgG1/IgG2a responses and higher hemagglutination inhibition titers compared to the reverse order (P-R regimen) [62].
Transcriptomic analysis revealed that heterologous prime-boost groups activated distinct immune response pathways depending on the immunization order. The R-P regimen showed enriched pathways for Th2 differentiation and CD8+ T-cell activation, while the P-R regimen activated mast cell and neutrophil degranulation pathways alongside helper T-cell diapedesis [62]. These findings demonstrate that the vaccination sequence qualitatively shapes the immune response, providing a strategic tool for personalization.
mRNA Vaccines as Immune Modulators in Oncology
Perhaps the most compelling evidence for personalized vaccinology comes from oncology applications, where SARS-CoV-2 mRNA vaccines have been shown to sensitize tumors to immune checkpoint blockade (ICB) [18]. In preclinical models, SARS-CoV-2 mRNA vaccines induced substantial increases in type I interferon, enabling innate immune cells to prime CD8+ T cells that target tumor-associated antigens [18]. This effect required concomitant ICB treatment for maximal efficacy in immunologically cold tumors, which responded by increasing PD-L1 expression [18].
Remarkably, analysis of patient cohorts revealed that receipt of SARS-CoV-2 mRNA vaccines within 100 days of initiating ICB was associated with significantly improved median overall survival in both non-small cell lung cancer (20.6 vs. 37.3 months) and melanoma (26.67 months vs. unmet) [18]. This benefit was similar among patients with immunologically cold tumors, suggesting that mRNA vaccines can fundamentally reset the tumor microenvironment to overcome resistance mechanisms [18]. These findings position clinically available mRNA vaccines as potent immune modulators capable of sensitizing tumors to immunotherapy, representing an unexpected form of personalization.
Experimental Methodologies: Monitoring and Adjusting for Heterogeneous Responses
Comprehensive Immune Monitoring Techniques
Accurate assessment of immune responses is fundamental to personalized vaccinology. The complex dynamics of vaccine immunity necessitate sophisticated monitoring approaches that capture both humoral and cellular parameters across multiple timepoints.
Table 2: Immune Monitoring Techniques for Personalized Vaccinology
| Technique | Parameters Measured | Application in Vaccinology | Considerations |
|---|---|---|---|
| Plaque Reduction Neutralization Test (PRNT) | Neutralizing antibody titers | Gold standard for functional humoral immunity | Technically demanding, requires BSL-2/3 facilities |
| ELISpot | Antigen-specific T cells (IFN-γ production) | Cellular immune response quantification | High sensitivity, single-cell resolution |
| Multiparametric Flow Cytometry | Immune cell phenotyping, intracellular cytokines | Comprehensive cellular immunity profile | Requires fresh cells, complex panel design |
| Multiplex Bead Assays | Cytokine/chemokine profiles | Systemic inflammatory environment | High-throughput, minimal sample volume |
| scRNA-seq | Transcriptomic profiles of individual cells | Unbiased immune response characterization | Expensive, complex computational analysis |
| Whole Blood Stimulation | Combined humoral and cellular responses | Preservation of physiological context | Minimal processing, clinical practicality |
Longitudinal studies are essential for capturing the temporal dynamics of vaccine responses, which typically feature rapid innate immune activation (hours to days), followed by peak adaptive responses (weeks), and eventual contraction leaving memory populations [60]. The integration of these multidimensional data types through advanced analytical approaches enables the identification of immune signatures predictive of vaccine efficacy and the detection of suboptimal responses requiring intervention.
Experimental Workflow for Personalized Vaccine Development
The following diagram illustrates a comprehensive experimental workflow for developing personalized vaccine strategies that account for heterogeneous immune responses:
Diagram 1: Experimental workflow for developing personalized vaccine strategies
This workflow begins with the identification of populations exhibiting heterogeneous responses to existing vaccines, proceeds through comprehensive immune profiling and mechanistic studies to identify key response determinants, and culminates in the development and validation of personalized vaccine approaches that can overcome response limitations.
Key Research Reagent Solutions
The following table details essential materials and reagents for investigating heterogeneous immune responses to mRNA vaccines and developing personalized approaches:
Table 3: Essential Research Reagents for Personalized mRNA Vaccinology
| Reagent Category | Specific Examples | Function/Application | Technical Notes |
|---|---|---|---|
| mRNA Constructs | Nucleoside-modified mRNA (m1Ψ) | Enhanced translation, reduced immunogenicity | Critical for balancing expression and reactogenicity [2] |
| CleanCap AG technology | Co-transcriptional capping (≥94% Cap-1) | Reduces innate sensing via RIG-I/IFIT1 [61] | |
| Delivery Systems | Ionizable LNPs (SM-102, ALC-0315) | mRNA encapsulation and cellular delivery | Primary driver of adjuvanticity [2] [30] |
| PEG-lipids | LNP stability, pharmacokinetic modulation | Influences reactogenicity and biodistribution | |
| Immune Assays | IFN-γ ELISpot | Antigen-specific T cell quantification | High sensitivity for cellular immunity [60] |
| Multiplex cytokine panels | Comprehensive inflammatory profiling | Captures innate immune activation [60] | |
| Cell Isolation | PBMC separation media | Peripheral blood mononuclear cell isolation | Foundation for ex vivo immune assays [60] |
| Magnetic bead cell separation | Specific immune cell population isolation | Enables cell-type-specific analyses |
Future Directions: Integrating Novel Technologies and Approaches
The future of personalized vaccinology lies in the integration of advanced technologies that enable precise modulation of vaccine responses based on individual immune characteristics. Several promising directions are emerging:
Advanced mRNA Design: Continued optimization of mRNA sequences through modified 5' cap structures, codon optimization, and untranslated region (UTR) engineering will further enhance translation efficiency and fine-tune immunogenicity [61]. The development of self-amplifying mRNA platforms offers the potential for dose-sparing and prolonged antigen expression, which may benefit populations with diminished responses to conventional mRNA vaccines [30].
Biomaterial-Based Delivery Innovations: Next-generation lipid nanoparticles with tissue-specific targeting capabilities and improved biodegradability profiles will enable more precise delivery of mRNA vaccines to desired cell populations while reducing systemic reactogenicity [61] [30]. The incorporation of pathogen-mimicking properties into delivery systems may further enhance immune activation in poor responders.
Systems Vaccinology and Predictive Modeling: The integration of multi-omics data through machine learning approaches will identify predictive signatures of vaccine response and enable the pre-vaccination identification of individuals likely to exhibit suboptimal immunity [60]. This predictive capability represents the cornerstone of truly personalized vaccinology, allowing for preemptive adjustment of vaccine formulations or regimens.
Combination Immunotherapy Strategies: The demonstrated ability of mRNA vaccines to sensitize tumors to immune checkpoint inhibitors suggests potential applications beyond infectious diseases [18]. Similar combination approaches may overcome response limitations in other clinical contexts, particularly in immunosenescent populations or individuals with compromised immunity.
Personalized vaccinology represents the inevitable evolution of immunization science, moving beyond uniform approaches to embrace the biological diversity of human populations. The strategic adjustment of vaccine strategies for heterogeneous immune responses requires deep understanding of the fundamental mechanisms connecting innate immune recognition of vaccine platforms to the development of adaptive immunity. mRNA vaccines, with their modular design and inherent immunomodulatory properties, offer an exceptionally flexible platform for personalization approaches.
By leveraging insights from heterologous vaccination strategies, advanced immune monitoring technologies, and mechanistic studies of vaccine immunology, researchers are developing increasingly sophisticated approaches to match vaccine formulations and regimens to individual immune characteristics. The ongoing integration of biomaterial engineering, systems biology, and predictive modeling will accelerate this transformation, ultimately enabling a future where vaccination strategies are as unique as the individuals receiving them.
From Bench to Bedside: Validating and Comparing Immune Outcomes
The rapid deployment of mRNA-LNP vaccines during the COVID-19 pandemic represented a paradigm shift in vaccinology, showcasing the potential of this platform to induce robust and protective immune responses. Despite their demonstrated efficacy, the precise immunological mechanisms through which mRNA-based vaccines initiate and coordinate innate and adaptive immunity remain incompletely defined. Single-cell transcriptomics has emerged as a powerful tool to dissect these complex immune dynamics at unprecedented resolution, particularly in the critical early sites of immune activation: the injection site and draining lymph nodes (dLNs). Understanding the initial chain of immune reactions elicited by mRNA vaccination is essential for optimizing future vaccine design and improving therapeutic outcomes [11] [20].
This technical guide examines how single-cell RNA sequencing (scRNA-seq) is revolutionizing our understanding of the spatial and temporal immune responses to mRNA vaccination. By profiling individual cells from these key anatomical locations, researchers can decrypt the precise cellular players, transcriptional programs, and signaling pathways that bridge innate sensing to adaptive protection. The integration of these data provides a comprehensive atlas of the immune symphony orchestrated by mRNA vaccines, from initial antigen sensing to the generation of lasting immunological memory [20] [63].
Fundamental Principles of mRNA Vaccine Immunology
mRNA vaccines function through a streamlined mechanism wherein delivered mRNA is translated into the target antigenic protein within host cells, predominantly antigen-presenting cells (APCs) at the injection site and in dLNs. This in situ antigen production mimics natural infection without associated risks, enabling robust immune activation. The vaccine mRNA is typically encapsulated within lipid nanoparticles (LNPs) that protect the nucleic acid and facilitate cellular uptake through endocytosis [20].
Once inside the cytoplasm, the mRNA is translated by host ribosomes into the encoded antigen, which undergoes processing and presentation on major histocompatibility complex (MHC) molecules. This direct antigen presentation, coupled with innate immune activation through pattern recognition receptors (PRRs), creates an inflammatory context that promotes dendritic cell maturation and migration to dLNs, where they prime naïve T cells and initiate adaptive immune responses [64] [20].
Critical to the success of this process is the careful balancing of immunostimulation and translation efficiency. Excessive innate immune activation can inhibit antigen production, while insufficient activation may fail to provide necessary co-stimulatory signals for adaptive immunity. Single-cell transcriptomics enables researchers to dissect these delicate balances at cellular resolution across different tissue compartments [65] [20].
Tissue Processing Methodologies
Injection Site Sampling and Processing
For comprehensive profiling of injection site responses, muscle tissue from the vaccination site must be collected at predetermined time points post-immunization. The following protocol has been optimized for murine models and can be adapted for other species:
- Tissue Collection: At designated intervals (e.g., 2, 16, 40 hours post-injection), resect the anterior thigh muscles where mRNA vaccines were administered.
- Mechanical and Enzymatic Dissociation: Mince tissue finely with surgical scissors, then digest using collagenase IV (1-2 mg/mL) and DNase I (100 μg/mL) in RPMI medium at 37°C for 30-45 minutes with continuous agitation.
- Single-Cell Suspension Preparation: Pass digested tissue through a 70μm cell strainer, then wash with cold PBS containing 2% fetal bovine serum (FBS).
- Cell Viability Maintenance: Use cold buffers throughout processing and consider adding viability dyes (e.g., propidium iodide) for dead cell exclusion during analysis.
- Immune Cell Enrichment (Optional): For rare cell populations, employ magnetic-activated cell sorting (MACS) with antibodies against CD45 for leukocyte enrichment [11].
This methodology has enabled researchers to profile 83,094 single cells from injection sites, identifying 22 distinct cell types including fibroblasts, endothelial cells, dendritic cells, monocytes, neutrophils, and T and B lymphocytes [11].
Draining Lymph Node Sampling Approaches
Precision-Cut Lymph Node Slices
For human LN studies where direct sampling is challenging, precision-cut LN slices provide an architecturally preserved ex vivo model system:
- LN Acquisition: Obtain healthy non-inflamed human LNs from surgical procedures (e.g., elective cholecystectomy).
- Tissue Preparation: Excise LNs from surrounding connective tissue and embed in low-melting-point agarose for stability during sectioning.
- Precision Sectioning: Use a vibratome to generate 300-μm-thick full-organ cross-sections. This thickness optimizes viability while preserving tissue architecture.
- Ex Vivo Culture: Maintain slices in complete RPMI medium supplemented with 10% FBS, L-glutamine, and antibiotics at 37°C with 5% CO₂.
- Stimulation and Analysis: Challenge slices with vaccine adjuvants (e.g., LMQ similar to AS01) for 20 hours before processing for scRNA-seq or other analyses [59].
This approach maintains viability of diverse LN cell populations, including innate lymphoid cells (ILCs), natural killer (NK) cells, monocytes, macrophages, and resident stromal populations, enabling study of their functional responses within native tissue context [59].
Ultrasound-Guided Fine Needle Aspiration (FNA)
For longitudinal sampling of human dLNs in clinical studies:
- LN Localization: Identify axillary LNs using high-frequency ultrasound imaging.
- Aspiration Procedure: Under ultrasound guidance, insert a 25-gauge needle into the LN cortex and apply suction while moving the needle through different trajectories.
- Sample Processing: Expel aspirates into cold PBS with 2% FBS, disaggregate by gentle pipetting, and filter through 40μm strainers.
- Cell Counting and Viability Assessment: Use automated cell counters with trypan blue exclusion to determine concentration and viability.
- Cryopreservation (Optional): Preserve cells in freezing medium (90% FBS, 10% DMSO) for batch analysis [63].
This minimally invasive approach enables serial sampling from the same LN over time, facilitating longitudinal studies of immune response evolution. It has been successfully used to track germinal center responses and T follicular helper cell dynamics for up to 6 months post-vaccination [63].
Single-Cell Library Preparation and Sequencing
After obtaining single-cell suspensions, the following workflow enables high-quality transcriptomic data:
- Cell Quality Control: Assess viability (>80% recommended) and ensure single-cell suspension without clumps.
- Cell Partitioning: Use droplet-based systems (10X Genomics Chromium) or plate-based methods (Smart-seq2) to capture individual cells.
- cDNA Synthesis and Amplification: Perform reverse transcription and PCR amplification following manufacturer protocols.
- Library Construction: Fragment amplified cDNA, add adapters, and incorporate sample indices.
- Quality Control and Sequencing: Assess library quality using Bioanalyzer, then sequence on appropriate platforms (Illumina NovaSeq, HiSeq) to sufficient depth (≥50,000 reads/cell recommended) [59] [11] [63].
Table 1: Key Considerations for Single-Cell RNA Sequencing Experiments
| Parameter | Recommendation | Rationale |
|---|---|---|
| Target Cell Recovery | 5,000-10,000 cells per sample | Balances cost with population representation |
| Sequencing Depth | 50,000-100,000 reads/cell | Captures sufficient transcript diversity |
| Viability Threshold | >80% | Redoves technical bias from dead cells |
| UMI Counts/Cell | >1,000 | Ensures adequate transcript capture |
| Gene Detection | >500 genes/cell | Indicates good RNA quality |
| Mitochondrial RNA | <20% | Minimizes technical bias from stressed cells |
Analytical Workflows for scRNA-seq Data
Primary Data Processing
The initial computational workflow transforms raw sequencing data into analyzable gene expression matrices:
- Demultiplexing and Alignment: Use cellranger (10X Genomics) or similar tools to assign reads to samples and align to reference genomes.
- UMI Counting: Quantify unique molecular identifiers (UMIs) to generate digital gene expression matrices.
- Quality Control Filtering: Remove cells with low UMI counts, few detected genes, or high mitochondrial content indicating poor viability.
- Normalization and Scaling: Apply methods like SCTransform or log-normalization to account for technical variability.
- Integration: Use Harmony, Seurat, or similar tools to combine datasets from multiple samples or time points while removing batch effects [59] [66] [63].
Cell Type Identification and Annotation
After dimensionality reduction (PCA, UMAP), cluster cells and assign identities:
- Unsupervised Clustering: Apply graph-based methods (Louvain, Leiden) to identify transcriptionally distinct cell populations.
- Marker Gene Identification: Find differentially expressed genes between clusters using Wilcoxon rank-sum tests.
- Cell Type Annotation: Reference canonical marker genes (e.g., CD3E for T cells, CD19 for B cells, CD68 for macrophages) and curated databases.
- Subpopulation Resolution: Iteratively subcluster to identify finer cellular subsets (e.g., CD4+ T cell helper subsets, monocyte subtypes) [59] [66].
Advanced Analytical Approaches
For deeper biological insights, implement these specialized analyses:
- Trajectory Inference: Use Monocle3, PAGA, or Slingshot to reconstruct cellular differentiation paths.
- Cell-Cell Communication: Apply tools like CellChat or NicheNet to infer signaling interactions between cell types.
- Gene Regulatory Networks: Utilize SCENIC or similar approaches to identify transcription factors driving cell states.
- Antigen Specificity Mapping: Integrate TCR/BCR sequencing with transcriptomic data to link clonality to cell phenotypes [63].
Key Findings from Single-Cell Transcriptomic Studies
Injection Site Responses
scRNA-seq of vaccination sites has revealed intricate cellular dynamics following mRNA-LNP administration. Studies profiling 83,094 single cells from injection sites identified two major axes of transcriptional responses:
Stromal Inflammatory Axis: Driven primarily by the LNP component, this response peaks around 16 hours post-injection and features induction of pro-inflammatory cytokines (IL-6, TNF, CCL2) in fibroblasts, endothelial cells, and mural cells. This creates a chemotactic gradient that recruits innate immune cells to the site [11].
Type I Interferon Axis: Specifically triggered by the mRNA component, this response is characterized by induction of interferon-stimulated genes (ISGs) like ISG15, OASL1, and IFIT3 in migratory dendritic cells. This response is critical for subsequent adaptive immunity [11].
Notably, mRNA vaccine transcripts show distinct cellular tropism at injection sites, with stromal cells (fibroblasts, endothelial cells, pericytes) and myeloid cells containing the highest abundance of spike mRNA at early time points (2 hours post-injection) [11].
Table 2: Key Cell Populations at mRNA Vaccine Injection Sites
| Cell Type | Abundance | Primary Function | Response to Vaccine |
|---|---|---|---|
| Fibroblasts | High | Structural support, inflammatory signaling | Major source of IL-6, CCL2; highly enriched with vaccine mRNA |
| Monocytes/Macrophages | Medium | Phagocytosis, antigen presentation | Inflammatory cytokine production; NLRP3 inflammasome activation |
| Migratory Dendritic Cells | Low | Antigen transport to dLNs | Type I IFN production; ISG expression; critical for T cell priming |
| Endothelial Cells | Medium | Vascular integrity, leukocyte recruitment | Adhesion molecule upregulation; chemokine production |
| Neutrophils | Variable | Early inflammatory response | Rapid recruitment; initial pathogen control |
Draining Lymph Node Responses
Single-cell profiling of dLNs has uncovered sophisticated cellular choreography following vaccination:
Early Innate Activation: Within hours of vaccination, monocytes and macrophages in dLNs directly respond to vaccine adjuvants through TLR4 and NLRP3 inflammasome activation, secreting IL-1β but not IL-18. This initial wave is TLR4-dependent [59].
Innate Lymphoid Cell Coordination: NK cells and other ILCs are indirectly activated by monocyte/macrophage-derived cytokines, subsequently secreting IFN-γ that signals downstream to B cells, bridging innate and adaptive immunity [59].
Stromal Cell Orchestration: Resident LN stromal populations, primed both directly and indirectly by vaccine components, play instrumental roles in mediating inflammatory cell recruitment, particularly neutrophils, through chemokine production [59].
CD4+ T Follicular Helper Cell Specialization: scRNA-seq of 1,277 spike-specific CD4+ T cells from human dLNs revealed remarkable heterogeneity in TFH populations, including germinal center TFH cells (CXCL13+CXCR5+BCL6+), IL-10+ TFH cells, cytotoxic TFH cells (GZMA+GZMK+), and effector TFH cells. These specialized subsets provide distinct forms of B cell help [63].
Longitudinal tracking of dLN responses through serial FNA has demonstrated that spike-specific TFH cells and germinal center B cells persist for at least 6 months post-vaccination, indicating sustained cellular immunity [63].
Signaling Pathways in mRNA Vaccine Immunity
The immune response to mRNA vaccines involves coordinated activation of multiple signaling pathways that bridge initial detection to adaptive immunity. The following diagrams visualize these key mechanisms.
Innate Immune Recognition Pathways
Diagram 1: Innate immune recognition pathways for mRNA vaccines. Vaccine components are detected by both endosomal and cytosolic pattern recognition receptors, triggering signaling cascades that produce inflammatory cytokines and type I interferons. These pathways shape subsequent adaptive immune responses [20].
Coordination of Lymph Node Immune Responses
Diagram 2: Coordination of draining lymph node responses to mRNA vaccination. The temporal cascade involves initial innate activation, intermediate ILC coordination, and eventual adaptive immunity. Cell-cell interactions and cytokine signaling bridge these phases [59] [63].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Single-Cell Studies of Vaccine Responses
| Reagent/Category | Specific Examples | Research Application | Technical Considerations |
|---|---|---|---|
| Single-Cell Platforms | 10X Genomics Chromium, BD Rhapsody, Smart-seq2 | Partitioning single cells for RNA-seq | 10X: high throughput, Smart-seq2: full-length transcripts |
| Cell Sorting Markers | CD45 (pan-immune), CD3 (T cells), CD19 (B cells), CD14 (monocytes), CD11c (dendritic cells) | Immune population enrichment | Use viability dyes; consider intracellular antigen staining |
| Cytokine Detection | IL-1β, IL-6, TNF, IFN-α/β, IFN-γ ELISAs; Luminex multiplex panels | Quantifying inflammatory mediators | Match sensitivity to expected concentrations |
| mRNA Vaccine Components | Nucleoside-modified mRNA (m1Ψ), ionizable lipids (ALC-0315), cholesterol, PEGylated lipids | Deconstructing vaccine mechanisms | Consider LNP size, PDI, encapsulation efficiency |
| Pathway Inhibitors | Anti-IFNAR antibodies, NLRP3 inhibitors (MCC950), TLR antagonists | Mechanistic studies of signaling pathways | Timing critical for in vivo interventions |
| Reference Databases | ImmGen, Blueprint Epigenome, Human Cell Atlas | Cell type annotation and validation | Use multiple references for robust annotation |
Single-cell transcriptomics has fundamentally advanced our understanding of mRNA vaccine immunology by revealing the precise cellular players and molecular programs that coordinate protection. The integration of data from injection sites and draining lymph nodes has illuminated the spatial and temporal dynamics of vaccine responses, from initial innate sensing to the generation of adaptive memory. These insights are already informing next-generation vaccine design, with optimization of mRNA modifications, LNP compositions, and dosing regimens to fine-tune immunogenicity and reactogenicity.
As single-cell technologies continue to evolve—with emerging capabilities in multi-omics, spatial transcriptomics, and computational integration—they promise to further decrypt the immune symphony orchestrated by mRNA vaccines. These advances will accelerate the development of not just improved vaccines for infectious diseases, but also mRNA-based therapies for cancer, autoimmune disorders, and other challenging conditions. The methodological framework presented here provides researchers with the tools to continue exploring this exciting frontier at the intersection of vaccinology and systems immunology.
The development of lipid nanoparticle (LNP)-delivered mRNA therapeutics represents a transformative advancement in vaccinology and molecular medicine. A critical understanding of this platform requires dissecting the individual contributions of the mRNA cargo and the LNP delivery vehicle to the overall immune response. This whitepaper synthesizes findings from recent murine and nonhuman primate (NHP) studies comparing LNP-mRNA with empty LNPs. Evidence confirms that the LNP itself functions as a potent adjuvant, primarily activating pro-inflammatory pathways, while the mRNA component, even when nucleoside-modified, induces a distinct Type I Interferon (IFN) response. However, significant interspecies differences in the magnitude, kinetics, and spatial distribution of these responses exist, influencing the translational predictive value of each model. This analysis provides a technical guide for selecting and interpreting in vivo models to deconvolute innate immune activation, which is essential for optimizing the safety and efficacy of next-generation mRNA therapeutics.
The LNP-mRNA platform's immunogenicity stems from the combined effects of its two core components: the ionizable lipid-based nanoparticle and the encapsulated mRNA. The innate immune system recognizes exogenous mRNA through various pattern recognition receptors (PRRs), including endosomal Toll-like receptors (TLR7/8) and cytosolic sensors like RIG-I and MDA5 [10]. Detection triggers signaling cascades that culminate in the production of Type I Interferons (IFN-α/β) and pro-inflammatory cytokines. Historically, nucleoside modification (e.g., N1-methylpseudouridine) and purification processes were implemented to render mRNA "immuno-silent," reducing innate sensing to enhance protein expression [67] [34].
Conversely, the LNP vehicle is not a passive carrier. Empty LNPs (devoid of mRNA) exhibit intrinsic adjuvant activity and reactogenicity. Ionizable lipids, a critical LNP component, can structurally resemble lipid A from lipopolysaccharide (LPS), enabling activation of the TLR4/MyD88 signaling pathway. This activation drives the production of pro-inflammatory cytokines such as IL-6, IL-1β, and TNF-α [68] [10]. This inherent immunogenicity of the LNP is a double-edged sword; while it provides necessary adjuvantation for vaccines, it can also contribute to adverse effects and inhibit mRNA translation, which is particularly problematic for protein-replacement therapies [68].
Therefore, a precise understanding of the platform requires a comparative approach using empty LNPs as a control to isolate the immunogenic contribution of the delivery vehicle from that of the mRNA cargo. The following sections detail the distinct innate immune profiles elicited by each component and highlight the critical differences observed between murine and NHP models.
Distinct Innate Immune Profiles of LNP and mRNA Components
LNP-Driven Immunity: The Stromal Pro-Inflammatory Axis
A key finding from single-cell transcriptomic studies in mice is that empty LNPs primarily drive a strong pro-inflammatory response in stromal cells at the injection site, including fibroblasts, endothelial cells, and pericytes [11]. This response is characterized by the upregulation of genes such as Il6, Tnf, and Ccl2 [11].
The mechanism behind this response often involves TLR4 activation. Research has demonstrated that the ionizable lipids in LNPs can activate the TLR4-MyD88 signaling axis. This pathway is essential for initiating reactogenic signals, pro-inflammatory gene expression, and physiological outcomes like reduced food intake and body weight in mice—classic metrics of sickness behavior [68]. The critical role of this pathway was confirmed using gene ablation studies, where MyD88-/- mice showed ablated reactogenicity, and the TLR4 inhibitor TAK-242 effectively mitigated LNP-driven inflammatory responses [68].
mRNA-Driven Immunity: The Type I Interferon Axis
In contrast to the LNP-driven pro-inflammatory response, the mRNA component is specifically responsible for inducing a robust Type I Interferon (IFN) response. This response is particularly evident in migratory Dendritic Cells (mDCs) at the injection site and draining lymph nodes (dLNs) [11]. This IFN response is dependent on signaling through the Interferon-α/β receptor (IFNAR) [67].
Notably, this response occurs even with nucleoside-modified mRNA, previously considered "immuno-silent" based on in vitro data [67] [11]. In vivo studies in mice show that LNP-mRNA vaccination leads to rapid IFNAR-dependent activation of dendritic cells, recruitment of monocytes to dLNs, and systemic cytokine responses [67]. A pivotal discovery is that injection site fibroblasts are highly enriched with delivered mRNA and are a primary source of IFN-β following vaccination [11]. This IFN-β production is crucial for inducing a unique population of migratory DCs high in IFN-stimulated genes (mDC_ISGs) and for promoting subsequent cellular immunity [11].
Table 1: Comparative Innate Immune Profiles of LNP and mRNA Components
| Immune Feature | Primary Eliciting Component | Key Signaling Molecules/Pathways | Primary Responding Cells |
|---|---|---|---|
| Pro-inflammatory Axis | Lipid Nanoparticle (LNP) | TLR4, MyD88, IL-6, IL-1β, CCL2 | Stromal cells (fibroblasts, endothelial), monocytes |
| Type I Interferon Axis | mRNA | IFNAR, RIG-I/MDA5, IFN-β, ISGs (e.g., Isg15, Oasl1) | Migratory Dendritic Cells (mDCs), fibroblasts |
Integrated Signaling Pathways
The following diagram synthesizes the primary innate immune signaling pathways triggered by LNP and mRNA components, as identified in the cited research.
Diagram 1: Innate immune signaling pathways activated by LNP-mRNA vaccine components. The LNP component (yellow) primarily activates the TLR4-MyD88-NF-κB pathway, driving pro-inflammatory cytokine production. The mRNA component (green) is sensed by cytosolic PRRs, leading to MAVS/IRF3 activation and Type I Interferon production, which acts via IFNAR to induce ISG expression.
Comparative Analysis of Murine and NHP Models
Translating findings from murine models to primates is a critical step in preclinical development. Comparative studies reveal that while the fundamental axes of immune activation are conserved, significant differences in the magnitude, kinetics, and distribution of responses exist.
Vaccine Distribution and Kinetics
The biodistribution and persistence of LNP-mRNA vaccines are highly dependent on the LNP formulation and the species. A comparative imaging study in mice and NHPs using two different ionizable lipids (MC3 and DOG-IM4) demonstrated starkly different distribution patterns [69].
In mice, MC3 LNPs (114 nm) showed rapid migration from the injection site to the draining lymph nodes (dLNs) within 6 hours. In contrast, DOG-IM4 LNPs (184 nm) persisted at the injection site for up to seven days, with only slow, limited trafficking to the dLNs [69]. This suggests that LNP composition and size can dramatically alter in vivo kinetics, a factor that must be considered when designing and interpreting animal studies.
Magnitude of Innate Immune and Physiological Responses
NHPs often exhibit a heightened innate immune response compared to mice. A barcoded LNP screening study found that the same pool of LNPs led to significantly higher transfection efficiency (aVHH+ cells) in 15 of 18 matched cell types in NHPs compared to mice [70].
Furthermore, physiological responses can differ. In NHPs, MC3 LNP-mRNA vaccination induced a transient elevation in rectal temperature and a significant increase in blood neutrophil and monocyte counts, alongside a sharp peak in serum IL-6, IL-1RA, IL-15, and CCL2 one day after injection [69]. These robust systemic cytokine responses were more pronounced than those typically observed in murine models, highlighting the NHP's value for assessing reactogenicity [69].
Impact on Adaptive Immunity
Studies in mice have revealed that the innate immune response can directly modulate adaptive immunity. The mRNA-induced IFNAR signaling, while activating innate immunity, can paradoxically attenuate the adaptive immune response. One study demonstrated that a brief, transient blockade of IFNAR signaling at the time of immunization significantly enhanced the frequencies of antigen-specific CD8+ T cells and elevated titers of antigen-specific antibodies [67]. This suggests that the Type I IFN response, while critical for initiating immunity, must be carefully regulated to achieve optimal vaccine efficacy.
Table 2: Key Differences in Murine and NHP Responses to LNP-mRNA Vaccines
| Parameter | Murine Models | Nonhuman Primate (NHP) Models | Translational Implication |
|---|---|---|---|
| LNP-mRNA Delivery Efficiency | Variable; model-dependent [69] | Generally higher across multiple tissues and cell types [70] | NHP data may better predict human delivery efficiency. |
| Innate Immune Kinetics | Rapid (hours to days); well-characterized at single-cell level [11] | Similar kinetics but often greater magnitude of cytokine response [69] | NHPs are critical for assessing systemic reactogenicity. |
| Physiological Response (e.g., Fever) | Measured as sickness behavior (e.g., weight loss) [68] | Directly measurable fever and acute phase responses [69] | NHP models more accurately capture human clinical reactogenicity. |
| Spatial Distribution | Highly dependent on LNP formulation (e.g., MC3 vs. DOG-IM4) [69] | Understudied, but imaging studies are feasible [69] | Formulation optimization in mice may not directly translate. |
| IFN-β Role in T-cell Immunity | Clearly demonstrated as critical; blockade enhances response [67] [11] | Inferable but less directly tested | Mechanistic insights from mice can inform NHP study design. |
Experimental Protocols for Component Deconvolution
Murine Model for Isolating mRNA vs. LNP Effects
Objective: To dissect the individual contributions of the LNP vehicle and mRNA cargo to the innate and adaptive immune response.
Key Reagents:
- Test Articles: LNP-encapsulated, nucleoside-modified mRNA (LNP-mRNA); Empty LNP (eLNP); Phosphate-Buffered Saline (PBS) control.
- mRNA Controls: Include mRNAs encoding different antigens and a non-coding mRNA sequence to control for antigen-specific effects [67].
- IFNAR Blocking: Anti-IFNAR monoclonal antibody (e.g., clone I-401-100 from Leinco Technologies) [67].
Methodology:
- Animal Models: Use female C57BL/6J (wild-type) and IFNAR-/- mice (age 6-8 weeks) [67].
- Immunization: Administer via intramuscular (i.m.) injection into the hind leg. A typical dose is 5 µg of LNP-mRNA or an equivalent volume of eLNP [67].
- IFNAR Blockade: Inject mice intraperitoneally with 2.5 mg of anti-IFNAR mAb 24 hours prior to and 24 hours post-immunization [67].
- Sample Collection:
- Early Time Points (6-48 hours): Analyze injection site (muscle) and draining lymph nodes (dLNs) for:
- Single-cell RNA sequencing (scRNA-seq) to identify differential gene expression and cell population changes [11].
- Flow Cytometry to quantify immune cell recruitment (monocytes, neutrophils, DCs) and activation.
- Cytokine Analysis (e.g., ELISA/MSD) to measure systemic levels of IFN-β, IL-6, etc. [67] [69].
- Early Time Points (6-48 hours): Analyze injection site (muscle) and draining lymph nodes (dLNs) for:
- Late Time Points (1-4 weeks): Assess adaptive immunity by measuring antigen-specific CD8+ T cells (via intracellular cytokine staining or tetramer staining) and antigen-specific antibody titers (via ELISA) [67].
NHP Model for Biodistribution and Systemic Reactogenicity
Objective: To evaluate the biodistribution, persistence, and systemic innate immune response of LNP-mRNA formulations in a translational model.
Key Reagents:
- Barcoded LNPs: A pool of LNPs with distinct chemical compositions, each carrying a unique DNA barcode and the same reporter mRNA (e.g., aVHH mRNA) [70].
- Imaging Agents: AF647-labeled mRNA or LNPs for in vivo tracking [69].
Methodology:
- Animal Models: Use purpose-bred NHPs or ethically sourced end-of-life (EoL) NHPs to minimize animal use [70].
- Administration: Administer the LNP pool intravenously (for biodistribution) or intramuscularly (for vaccine studies) at a standard nucleic acid dose (e.g., 0.5 mg/kg) [70] [69].
- In Vivo Imaging:
- Use fluorescence imaging (for mice) and PET/CT combined with near-infrared (NIR) imaging (for NHPs) to track LNP-mRNA migration from the injection site to dLNs over days [69].
- Systemic Monitoring:
- Tissue Analysis:
- At endpoint (e.g., 24 hours post-injection), harvest tissues (liver, spleen, bone marrow, blood, dLNs).
- Digest tissues into single cells and use FACS to isolate transfected (aVHH+) cells.
- Sequence the DNA barcodes from sorted cells to determine the functional delivery profile and rank-order potency of each LNP in the pool across different tissues and cell types [70].
The following diagram illustrates the workflow for a barcoded LNP study in NHPs.
Diagram 2: Workflow for barcoded LNP screening in vivo. A pool of LNPs, each with a unique DNA barcode, is administered to animals. After tissue harvest, transfected (aVHH+) cells are sorted and the barcodes are sequenced to quantify the delivery efficiency of each LNP formulation across different cell types.
The Scientist's Toolkit: Key Research Reagents and Models
Table 3: Essential Reagents and Models for LNP-mRNA Immune Profiling
| Category | Specific Reagent / Model | Function / Application | Key Findings Enabled |
|---|---|---|---|
| In Vivo Models | C57BL/6J & BALB/c mice | Standard model for initial mechanistic studies. | Deconvolution of LNP vs. mRNA immunity; role of IFNAR [67] [11]. |
| IFNAR-/- mice | Model to dissect Type I Interferon signaling. | Confirmed IFN-β's critical role in shaping cellular immunity [67] [11]. | |
| MyD88-/- & TRIF-/- mice | Model to dissect TLR4 signaling pathways. | Identified MyD88 as essential for LNP reactogenicity and sickness behavior [68]. | |
| End-of-Life (EoL) NHPs | Ethically sourced model for translational screening. | Enabled high-throughput LNP screening with minimal animal loss [70]. | |
| Critical Reagents | Anti-IFNAR mAb (e.g., I-401-100) | Transient blockade of Type I IFN signaling. | Enhanced antigen-specific T & B cell responses by blocking early IFN [67]. |
| TLR4 Inhibitor (TAK-242) | Pharmacological inhibition of TLR4 signaling. | Mitigated LNP-induced reactogenicity and pro-inflammatory cytokines [68]. | |
| Barcoded LNP Libraries | Pooled screening of LNP formulations in vivo. | Quantified delivery efficiency and identified top performers across species [70]. | |
| scRNA-seq & Bioinformatics | High-resolution transcriptomic profiling. | Identified fibroblast IFN-β production and mDC_ISG populations [11]. |
The comparative analysis of murine and NHP models unequivocally demonstrates that both the LNP vehicle and the mRNA cargo are integral to the overall immunogenicity of the platform. The LNP drives a primarily pro-inflammatory response via the TLR4/MyD88 axis, while the mRNA induces a robust, IFNAR-dependent Type I Interferon response, even in its nucleoside-modified form.
The choice of animal model is critical. Murine models provide unparalleled power for mechanistic dissection through the use of knockout strains and detailed spatial transcriptomics. NHP models, conversely, are indispensable for assessing the translational potential of formulations, offering superior predictive value for human biodistribution, systemic reactogenicity, and adaptive immunogenicity.
Future research should focus on leveraging these comparative insights to design smarter vaccines and therapeutics. Strategies such as incorporating TLR agonists like TLR7/8 into LNPs can enhance Th1-skewed adaptive immunity for vaccines against cancer or intracellular pathogens [34]. Conversely, for protein-replacement therapies, strategies that minimize LNP reactogenicity (e.g., using TLR4 inhibitors or novel ionizable lipids) and modulate IFN responses will be key to improving safety and efficacy. A rational design that exploits or suppresses these distinct immune pathways, validated across appropriate animal models, will unlock the full potential of the LNP-mRNA platform.
The clinical success of mRNA-Lipid Nanoparticle (LNP) vaccines has fundamentally transformed vaccinology, yet the precise immunological mechanisms that link initial innate sensing to durable adaptive protection remain a central focus in immunology research. A comprehensive understanding of these mechanisms is critical for the rational design of next-generation vaccines and therapeutics. This whitepaper synthesizes current research on the innate immune biomarkers elicited by mRNA-based vaccines and their direct role in shaping subsequent antigen-specific cellular and humoral immunity. Framed within the broader context of innate immune responses to exogenous mRNA delivery, this analysis provides drug development professionals with a detailed examination of key correlates of protection, experimental methodologies for their assessment, and the complex signaling networks that coordinate these responses.
Key Innate Biomarkers and Their Adaptive Correlates
The initial host response to mRNA-LNP vaccination is characterized by a predictable cascade of innate immune activation. This response can be quantified through specific cellular and molecular biomarkers that serve as early indicators of the ensuing adaptive immune quality and magnitude. The following table synthesizes key innate biomarkers and their established adaptive immune correlates based on recent preclinical and clinical findings.
Table 1: Key Innate Biomarkers and Their Link to Adaptive Immune Outcomes
| Innate Biomarker | Cell Source / Origin | Kinetics Post-Vaccination | Linked Adaptive Outcome | Experimental Evidence |
|---|---|---|---|---|
| Type I Interferons (IFN-α/β) [11] [49] | Fibroblasts, Dendritic Cells [11] | Peaks within 24 hours [11] | Enhancement: Promotes antigen-specific CD8+ T cells and antibody responses; Paradoxical Effect: Can be attenuating if signaling is excessive [49] | Blocking IFNAR signaling in vivo enhances antigen-specific T cells and antibodies [49] |
| IFN-Stimulated Genes (ISGs) [11] | Migratory Dendritic Cells (mDCs) [11] | Detectable by 16 hours [11] | Induction of mDC_ISG population; Critical for robust cellular immunity [11] | Single-cell RNA-seq identifies mDC_ISGs at injection site and dLNs; Depletion reduces T-cell responses [11] |
| Chemokine (MCP-1/CCL2) [24] | Myeloid Cells, Stromal Cells [11] | Elevated within 8 hours [24] | Recruitment of monocytes and T cells to lymphoid sites; Correlates with broader vaccine protection [24] | Higher MCP-1 in multi-antigen mRNA vaccines (mRNA-S+N) linked to broader protection against viral variants [24] |
| Inflammatory Cytokine (IL-6) [24] | Stromal Cells, Myeloid Cells [11] [24] | Elevated within 8 hours [24] | Associated with robust T follicular helper and germinal center B-cell reactions [11] | LNP-induced IL-6 is required for T-cell and B-cell responses; Correlates with antibody titers [11] [24] |
| Migratory Dendritic Cells (mDC_ISG) [11] | Draining Lymph Nodes [11] | Appears by 16 hours post-injection [11] | Antigen presentation and cross-presentation to naive T cells; Essential for cellular immune priming [11] | mDC_ISG induction is mRNA-specific; Ablation significantly decreases antigen-specific T-cell expansion [11] |
Mechanistic Insights: From Innate Sensing to Adaptive Priming
The connection between innate biomarkers and adaptive immunity is not merely correlative but is underpinned by defined mechanistic pathways. The biological trajectory begins with the delivery of mRNA-LNPs into the cytoplasm of target cells at the injection site, culminating in the generation of effector T cells and memory B cells.
Spatial Immunological Cascade
The following diagram illustrates the key spatial and temporal events in this cascade, from the muscle injection site to the draining lymph node.
Key Innate Signaling Pathways
The innate immune response is primarily triggered by the sensing of vaccine components by Pattern Recognition Receptors (PRRs). The diagram below details the major signaling pathways involved, highlighting the roles of both the mRNA and LNP components.
Detailed Experimental Protocols for Correlate Analysis
To reliably establish the links between innate and adaptive immunity, robust and detailed experimental methodologies are required. The following protocols are derived from seminal studies in the field.
Single-Cell Transcriptomic Profiling of the Injection Site
This protocol is designed to comprehensively map the cellular players and their transcriptional states at the site of mRNA-LNP administration [11].
- Animal Model: Female BALB/c mice.
- Immunization: Intramuscular injection of mRNA-LNP, empty LNP (control), or saline (control) in the anterior thigh muscle. Prime and boost shots with a 3-week interval.
- Tissue Harvest: Resect the injection site (anterior thigh muscles) at multiple time points post-injection (e.g., 2, 16, 40 hours).
- Single-Cell Suspension: Mechanically and chemically digest the muscle tissue to create a single-cell suspension.
- Library Construction & Sequencing: Construct single-cell RNA sequencing libraries using a platform such as 10x Genomics. Target a depth of ~50,000 cells per sample.
- Bioinformatic Analysis:
- Cell Type Identification: Cluster cells based on gene expression profiles using Seurat or Scanpy and annotate cell types with canonical markers (e.g., Pecam1 for endothelial cells, Pdgfra for fibroblasts, Itgam for myeloid cells).
- Differential Expression: Identify differentially expressed genes (DEGs) for each cell type between vaccinated and control groups.
- Spike mRNA Tracking: Map sequencing reads to a custom reference (e.g., SARS-CoV-2 spike open reading frame) to identify and quantify which cells have taken up the vaccine mRNA.
- Pathway Analysis: Perform gene set enrichment analysis (GSEA) on DEG lists to identify upregulated pathways (e.g., "Interferon Alpha Response," "Inflammatory Response").
Functional Validation of IFN-β Signaling
This protocol uses antibody-mediated blockade to establish the causal role of a specific innate biomarker in shaping adaptive immunity [11] [49].
- In Vivo Blockade: Administer an anti-IFNAR blocking antibody or an isotype control antibody to mice via intraperitoneal injection. Treatment should be timed to coincide with vaccination (e.g., 1 hour before and 24 hours after mRNA-LNP injection).
- Control Groups: Include groups that receive:
- mRNA-LNP + anti-IFNAR.
- mRNA-LNP + isotype control.
- Empty LNP.
- Saline.
- Sample Collection:
- Innate Phase (24-48h): Analyze injection site tissue (via scRNA-seq or bulk PCR for ISGs) and serum (via LEGENDplex bead-based assay for cytokines like IL-6, MCP-1).
- Adaptive Phase (≥2 weeks): Collect spleen and blood.
- Immune Readouts:
- Cellular Immunity: Perform IFN-γ ELISpot assay or intracellular cytokine staining (ICS) on splenocytes stimulated with antigen-derived peptides to quantify antigen-specific T cells.
- Humoral Immunity: Measure antigen-specific antibody titers (total IgG, subclasses) and neutralizing antibody capacity using ELISA and PRNT assays, respectively.
Longitudinal Analysis of Systemic and Local Immune Responses
This protocol correlates early systemic innate signals with later adaptive protection, particularly useful for evaluating combination vaccines [24].
- Vaccination Groups: Immunize mice with single-antigen (mRNA-S) and multi-antigen (mRNA-S+N) mRNA-LNP vaccines, including empty LNP controls.
- Innate Immune Sampling (Day 1):
- Serum: Collect blood 8 hours post-prime immunization. Use a bead-based multiplex immunoassay (e.g., BioLegend LEGENDplex) to quantify serum cytokines/chemokines (IL-6, MCP-1, IFNs).
- Lymph Nodes: Harvest draining lymph nodes 24 hours post-immunization. Create single-cell suspensions for immunophenotyping by flow cytometry to assess activation/maturation markers on dendritic cells (e.g., CD80, CD86, MHC-II) and NK cells.
- Adaptive Immune Sampling (Weeks 2-5):
- Tissues: Collect spleen and lung two weeks post-booster for RNA-Seq analysis.
- Transcriptomics: Perform bulk RNA-Seq and analyze differential gene expression and pathway enrichment (e.g., T cell, B cell, cytokine signaling pathways) to compare the breadth of immune activation between vaccine groups.
- Functional Assays: As in Protocol 4.2, conduct ELISpot and neutralization assays to confirm the functional superiority linked to the stronger innate signature.
The Scientist's Toolkit: Essential Reagents and Models
The following table catalogues critical reagents, model systems, and tools employed in the cited research to dissect innate-adaptive immune correlates.
Table 2: Essential Research Tools for Investigating mRNA Vaccine Immunity
| Tool / Reagent | Specific Example(s) | Research Application | Key Function in Studies |
|---|---|---|---|
| Ionizable LNPs | ALC-0315, DLin-MC3-DMA [24] | Vaccine Formulation | Delivers mRNA into cytoplasm; provides intrinsic adjuvanticity by activating innate immune cells [24]. |
| Modified mRNA | 1-methylpseudouridine (m1Ψ) [24] [23] | Antigen Encoding | Reduces excessive innate immune recognition, enhances translational efficiency and stability of mRNA [24] [23]. |
| Single-Cell RNA Seq | 10x Genomics Platform [11] | Transcriptional Profiling | Unbiased identification of novel cell states (e.g., mDC_ISGs) and cellular tropism of mRNA at injection site [11]. |
| Anti-IFNAR Antibody | MAR1-5A3 (clone) [49] | Functional Blocking | Causally links Type I IFN signaling to adaptive immune outcomes by transiently blocking the IFNAR receptor in vivo [49]. |
| LEGENDplex Assay | Mouse Inflammation Panel [24] | Cytokine Quantification | Multiplexed, high-sensitivity measurement of key serum biomarkers (e.g., IL-6, MCP-1) following vaccination [24]. |
| Humanized Mouse Models | NSG or NOG mice engrafted with human immune cells [71] | Preclinical Testing | Provides an in vivo model to study human-specific immune responses to mRNA vaccines in a controlled system [71]. |
| 3D Organoid Co-cultures | Patient-derived tumor organoids + autologous immune cells [71] | Human Ex Vivo Modeling | Recapitulates human tumor microenvironment to test mRNA vaccine-induced T cell reactivity in a patient-specific context [71]. |
The rigorous dissection of clinical correlates linking innate biomarkers to adaptive immune protection is fundamental for advancing the mRNA vaccine platform. Evidence now solidly implicates specific innate parameters—including IFN-β from injection-site fibroblasts, the subsequent ISG signature in dendritic cells, and LNP-driven inflammatory cytokines—as non-redundant drivers of the quantity and quality of the antigen-specific T and B cell response. However, this relationship is complex, as evidenced by the dual role of IFNAR signaling, which can be both essential and potentially suppressive. Future research must focus on refining these correlates in diverse human populations, leveraging single-cell technologies and functional genetic screens to build predictive models of vaccine efficacy. This deeper mechanistic understanding will ultimately enable the rational design of next-generation mRNA therapeutics with tailored immunogenicity, maximal efficacy, and superior safety profiles for a broad range of applications beyond infectious diseases.
Conclusion
The innate immune response to exogenous mRNA is a finely orchestrated but complex process that dictates the success of mRNA therapeutics. Key takeaways reveal that while the LNP carrier provides essential adjuvant activity, the mRNA itself is a critical driver of IFNAR-dependent pathways, with a nuanced role that can either potentiate or attenuate the desired adaptive immunity. Future progress hinges on the continued refinement of mRNA chemistry and LNP formulations, guided by high-resolution computational and omics technologies. The strategic modulation of this innate 'symphony'—particularly through transient IFNAR blockade or the use of translation boosters—promises to unlock a new generation of highly efficacious, well-tolerated, and rapidly adaptable mRNA vaccines and drugs for a broad spectrum of diseases.
References
- 1. Pattern recognition receptors: function, regulation and ... [https://www.nature.com/articles/s41392-025-02264-1]
- 2. Innate immune mechanisms of mRNA vaccines - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9641982/]
- 3. Sensing of RNA Viruses: a Review of Innate Immune ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3302314/]
- 4. Pattern Recognition and Signaling Mechanisms of RIG-I ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4107945/]
- 5. The molecular mechanism of RIG‐I activation and signaling [https://pmc.ncbi.nlm.nih.gov/articles/PMC9293153/]
- 6. RIG-I-like receptors: their regulation and roles in RNA ... [https://www.nature.com/articles/s41577-020-0288-3]
- 7. Regulation of RIG-I-like receptor-mediated signaling [https://www.nature.com/articles/s41423-020-00602-7]
- 8. mRNA vaccines for infectious diseases: principles, delivery ... [https://www.nature.com/articles/s41573-021-00283-5]
- 9. The mRNA component of LNP-mRNA vaccines triggers ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1670350/full]
- 10. Immunogenicity of lipid nanoparticles and its impact on the ... [https://www.nature.com/articles/s12276-023-01086-x]
- 11. Innate immune responses against mRNA vaccine promote ... [https://www.nature.com/articles/s41467-024-51411-9]
- 12. Straight to the point: targeted mRNA-delivery to immune cells ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10711611/]
- 13. Innate immune responses to three doses of the BNT162b2 ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2022.947320/full]
- 14. Innate immune responses against mRNA vaccine promote ... [https://pubmed.ncbi.nlm.nih.gov/39191748/]
- 15. Interferon and immunity: the role of microRNA in viral ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1567459/full]
- 16. A comparative computational analysis of IFN-alpha ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12500084/]
- 17. Unraveled role of TLR7-mediated interferon signaling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12540318/]
- 18. SARS-CoV-2 mRNA vaccines sensitize tumours to immune ... [https://www.nature.com/articles/s41586-025-09655-y]
- 19. Real-time visualization of STAT activation in live cells using ... [https://www.nature.com/articles/s41589-025-02012-0]
- 20. Decrypting the Immune Symphony for RNA Vaccines [https://www.mdpi.com/2076-393X/13/8/882]
- 21. mRNA vaccines contribute to innate and adaptive immunity ... [https://www.sciencedirect.com/science/article/abs/pii/S0142961224001625]
- 22. mRNA Vaccine Nanoplatforms and Innate Immunity - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10820759/]
- 23. Progress and prospects of mRNA-based drugs in pre- ... [https://www.nature.com/articles/s41392-024-02002-z]
- 24. Innate and Adaptive Immune Parameters following mRNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11125668/]
- 25. Innate and Adaptive Immune Parameters following mRNA ... [https://www.mdpi.com/2076-393X/12/5/543]
- 26. mRNA medicine: Recent progresses in chemical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12311907/]
- 27. Mechanisms of innate and adaptive immunity to the Pfizer ... [https://www.nature.com/articles/s41590-022-01163-9]
- 28. Synergistic effect of nucleoside modification and ionizable ... [https://www.nature.com/articles/s41541-025-01263-1]
- 29. ZUGC-RNA degradation generates immunosuppressor to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11838226/]
- 30. mRNA Vaccines: Current Applications and Future Directions [https://pmc.ncbi.nlm.nih.gov/articles/PMC12572956/]
- 31. Endosomal escape and current obstacles in ionizable lipid ... [https://www.sciencedirect.com/science/article/pii/S0378517325011007]
- 32. Design Strategies for Novel Lipid Nanoparticle for mRNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12497691/]
- 33. Artificial intelligence-driven rational design of ionizable ... [https://www.nature.com/articles/s41467-024-55072-6]
- 34. Integration of TLR7/8 agonists into lipid nanoparticles ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12629014/]
- 35. Lipid Nanoparticle (LNP) News Brief: March 17-24, 2025 [https://www.precigenome.com/post/lipid-nanoparticle-lnp-drug-delivery-advances-march-17-24-2025?srsltid=AfmBOooKKooKGMdNFNTkw8ey2BJk5U9vuVIV2CTdo0chFaC1cWd7r4G8]
- 36. a comprehensive review of mRNA vaccine technology and ... [https://virologyj.biomedcentral.com/articles/10.1186/s12985-025-02645-6]
- 37. Fine-Tuning mRNA Structure for Better mRNA Therapies [https://blog.genewiz.com/fine-tuning-mrna-structure-for-better-mrna-therapies]
- 38. mRNA vaccine sequence and structure design ... [https://www.sciencedirect.com/science/article/pii/S0021925824025171]
- 39. Algorithm for optimized mRNA design improves stability ... [https://www.nature.com/articles/s41586-023-06127-z]
- 40. CN108368028A - Novel Lipid and Lipid Nanoparticle ... [https://patents.google.com/patent/CN108368028A/en]
- 41. Comparison of immunogenicity and protection efficacy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12478056/]
- 42. Unleashing the potential of mRNA: Overcoming delivery ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11883111/]
- 43. Harnessing the Loop: The Perspective of Circular RNA ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12389747/]
- 44. Drug delivery systems for RNA therapeutics [https://www.nature.com/articles/s41576-021-00439-4]
- 45. Signaling by Type I Interferons in Immune Cells [https://pmc.ncbi.nlm.nih.gov/articles/PMC11049350/]
- 46. Interferon Alpha/Beta Receptor 2 drugs in development, 2024 [https://www.pharmaceutical-technology.com/data-insights/interferon-alpha-beta-receptor-2-drugs-in-development/]
- 47. The Dual Nature of Type I and Type II Interferons - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6141705/]
- 48. What are IFNAR-1 stimulants and how do they work? [https://synapse.patsnap.com/article/what-are-ifnar-1-stimulants-and-how-do-they-work]
- 49. The mRNA component of LNP-mRNA vaccines triggers ... [https://pubmed.ncbi.nlm.nih.gov/41169401/]
- 50. Type I interferon signaling induces melanoma cell-intrinsic ... [https://www.nature.com/articles/s41467-024-51496-2]
- 51. The how's and what's of vaccine reactogenicity [https://www.nature.com/articles/s41541-019-0132-6]
- 52. Controlling reactogenicity while preserving immunogenicity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12041602/]
- 53. Advax adjuvant formulations promote protective immunity ... [https://www.sciencedirect.com/science/article/abs/pii/S0264410X21002218]
- 54. The Strategies and Advances of mRNA Translation Booster [https://www.sciencedirect.com/science/article/pii/S1818087625000741]
- 55. Decrypting the Immune Symphony for RNA Vaccines - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12390541/]
- 56. Deep generative optimization of mRNA codon sequences ... [https://www.nature.com/articles/s41467-025-64894-x]
- 57. Artificial intelligence-guided design of lipid nanoparticles for ... [https://pubmed.ncbi.nlm.nih.gov/39658727/]
- 58. Artificial Intelligence-Driven Strategies for Targeted Delivery ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12389219/]
- 59. Ex vivo model of functioning human lymph node reveals role ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12284374/]
- 60. Monitoring Immune Responses to Vaccination: A Focus on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12030821/]
- 61. mRNA therapeutics: Transforming medicine through ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12590236/]
- 62. Analyzing immune responses to varied mRNA and protein ... [https://www.nature.com/articles/s41541-023-00684-0]
- 63. CD4+ T cells exhibit distinct transcriptional phenotypes in the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11627549/]
- 64. A Comprehensive Review of mRNA Vaccines - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9917162/]
- 65. Reducing cell intrinsic immunity to mRNA vaccine alters ... [https://www.sciencedirect.com/science/article/pii/S2162253123002639]
- 66. Single-Cell RNA Sequencing of Baseline Immune Profiles ... [https://www.mdpi.com/1422-0067/26/8/3494]
- 67. The mRNA component of LNP-mRNA vaccines triggers ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12568647/]
- 68. Lipid Nanoparticles Elicit Reactogenicity and Sickness ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11916992/]
- 69. Distinct dynamics of mRNA LNPs in mice and nonhuman ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11189915/]
- 70. Lipid nanoparticle screening in nonhuman primates with ... [https://www.nature.com/articles/s41587-025-02711-y]
- 71. Preclinical ex vivo and in vivo models to study immunotherapy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12574409/]