Engineering Lipid Nanoparticles for Precision mRNA Reprogramming: From Design to Clinical Translation
This article provides a comprehensive analysis of lipid nanoparticle (LNP) technology for delivering reprogramming mRNA, a revolutionary approach in therapeutics.
Engineering Lipid Nanoparticles for Precision mRNA Reprogramming: From Design to Clinical Translation
Abstract
This article provides a comprehensive analysis of lipid nanoparticle (LNP) technology for delivering reprogramming mRNA, a revolutionary approach in therapeutics. It explores the foundational principles of mRNA-LNP systems, detailing advanced engineering strategies for cell-specific targeting and enhanced efficacy. The content covers methodological innovations across diverse applications, including CAR-T cell engineering, immune modulation, and metabolic disease treatment. It further addresses critical challenges in delivery efficiency, tissue-specific targeting, and safety optimization, while evaluating performance through comparative studies and advanced characterization techniques. Aimed at researchers, scientists, and drug development professionals, this review synthesizes current advancements and future directions for realizing the full potential of mRNA-LNP reprogramming in precision medicine.
The Science of mRNA-LNP Reprogramming: Core Principles and Therapeutic Advantages
Lipid nanoparticles (LNPs) have emerged as the leading non-viral delivery platform for nucleic acid therapeutics, including the revolutionary application of delivering reprogramming messenger RNA (mRNA). The successful clinical deployment of LNP-based COVID-19 vaccines has accelerated interest in their use for more complex applications, such as the generation of induced pluripotent stem cells (iPSCs) [1] [2]. For reprogramming somatic cells like peripheral blood mononuclear cells (PBMCs) into iPSCs, mRNA encoding key transcription factors (e.g., Oct4, Sox2, Klf4, c-Myc) must be efficiently delivered into the cell's cytoplasm without genomic integration, a significant advantage over viral vectors [1]. The core structure of an LNP, which enables this efficient delivery, is a multi-component system composed of ionizable lipids, phospholipids, cholesterol, and PEG-lipids [3] [4]. Each component plays a distinct and critical role in the encapsulation, stability, delivery, and intracellular release of the reprogramming mRNA cargo. The precise formulation and understanding of these components are therefore paramount for developing effective and safe reprogramming protocols. This document provides detailed application notes and experimental protocols for the formulation and functional analysis of LNPs, specifically within the context of reprogramming mRNA research.
The Functional Roles of LNP Components
The following diagram illustrates the journey of an mRNA-loaded LNP from assembly to intracellular mRNA release, highlighting the functional roles of each lipid component at every stage.
Ionizable Lipids: The Engine of Endosomal Escape
Ionizable lipids are the cornerstone of modern LNP technology, serving as the primary determinant for efficient intracellular mRNA delivery [5] [6]. Their key characteristic is a pKa typically between 6.0 and 7.0, which allows them to be neutral at physiological pH (reducing toxicity) but to become positively charged in the acidic environment of the endosome (pH ~5.5-6.5) [3] [6]. This protonation triggers a critical structural change, enabling the lipid to interact with anionic endosomal phospholipids and form non-bilayer, cone-shaped structures that disrupt the endosomal membrane and facilitate the release of mRNA into the cytosol [5] [6].
Table 1: Classification and Properties of Ionizable Lipids for mRNA Delivery
| Lipid Type | Key Structural Features | Mechanism & Advantages | Example Lipids | Potency & Degradability Trade-off |
|---|---|---|---|---|
| Unsaturated | Linoleyl tails with cis double bonds [5] | Increased tendency to form non-bilayer phases; enhanced membrane disruption and payload release [5]. | DLin-MC3-DMA (MC3) [5] | High potency, but slow degradability can lead to accumulation [5]. |
| Multi-tail | Three or more hydrophobic tails [5] | Cone-shaped structure enhances endosome-disrupting ability; easily synthesized via combinatorial chemistry [5]. | 98N12-5, C12-200 [5] | High potency, but stable backbone may cause toxicity with repeated dosing [5]. |
| Biodegradable | Incorporation of ester or disulfide bonds [5] | Rapid clearance and improved tolerability; reduced side effects for repeated dosing [5]. | L319, 306-O12B [5] | Rapid hydrolysis can reduce delivery efficiency (activity-degradability tradeoff) [5]. |
| Branched-tail | Alkyl tails with methyl or other branches [5] | Enhanced protonation and cone-shaped structure; stronger endosomal escape and higher transfection [5]. | 306Oi10, FTT5 [5] | Slower degradation of secondary esters can maintain potency while improving safety [5]. |
Protocol 2.1.1: High-Throughput Screening of an Ionizable Lipid Library This protocol is adapted from combinatorial screening approaches used to identify lead ionizable lipids [5].
- Library Synthesis: Synthesize a library of ionizable lipids using a combinatorial reaction scheme (e.g., coupling alkyl amines with acrylate tails via Michael addition) [5].
- Microfluidic LNP Formulation: Use a microfluidic mixer (e.g., NanoAssemblr) to formulate LNPs with each library lipid. Hold the other components constant: phospholipid (DOPE or DSPC, 10 mol%), cholesterol (40 mol%), PEG-lipid (1.5 mol%), and a fixed N/P (nitrogen-to-phosphate) ratio [5] [7].
- In Vitro Potency Assay: Transfect a relevant cell line (e.g., HEK293T or HeLa) with LNPs encapsulating firefly luciferase (FLuc) mRNA. Measure luminescence 24 hours post-transfection to assess protein expression levels [8] [7].
- pKa Determination: Determine the apparent pKa of lead LNPs using a TNS (6-(p-Toluidino)-2-naphthalenesulfonic acid) fluorescence assay across a pH gradient (e.g., pH 4-10). LNPs with pKa between 6.0 and 7.0 are typically most effective [3].
- Lead Validation: Select top-performing lipids for in vivo validation in an animal model (e.g., mice) by measuring target protein expression in the liver or other target tissues after intravenous administration of FLuc mRNA LNPs [5].
Phospholipids: Structural "Helpers" with Active Functional Roles
Phospholipids are often termed "helper lipids," but they play an active role in defining LNP structure and function. They primarily contribute to the formation of the lipid bilayer that surrounds the LNP core, providing structural integrity [8] [9]. The choice of phospholipid significantly influences intracellular delivery and organ tropism. Phospholipids with a phosphoethanolamine (PE) headgroup, such as DOPE, adopt a conical molecular geometry that promotes the transition to an inverted hexagonal (HII) phase, thereby enhancing membrane fusion and endosomal escape [8] [9]. In contrast, phospholipids with a phosphocholine (PC) headgroup, such as DSPC, have a cylindrical shape that favors stable bilayer formation [9].
Table 2: Impact of Phospholipid Chemistry on LNP Performance
| Phospholipid | Head Group | Tail Saturation | Key Functional Contributions | Optimal Application Context |
|---|---|---|---|---|
| DSPC | PC (Phosphocholine) | Saturated (Stearoyl, C18:0) [8] | Enhances membrane stability; forms stable lamellar bilayers; standard in approved drugs/vaccines [8] [9]. | Formulations requiring high structural stability; standard component in Onpattro and COVID-19 vaccines [8]. |
| DOPE | PE (Phosphoethanolamine) | Unsaturated (Oleoyl, C18:1) [8] | Fusogenic; promotes inverted hexagonal (HII) phase; significantly enhances endosomal escape and mRNA translation [8] [9]. | Research applications where maximized transfection efficiency is critical; shown to boost delivery up to 4-fold in vivo [8]. |
| DOPC | PC (Phosphocholine) | Unsaturated (Oleoyl, C18:1) [9] | Provides fluid bilayer but less fusogenic than DOPE; can be used in SORT LNPs to modulate targeting [9]. | Basic LNP formulations; used in SORT LNP systems for organ-specific targeting [9]. |
| BMP | Anionic, two headgroups | Varies | Unique to endosomal membranes; its inclusion may enhance endosomal escape via membrane similarity [8]. | Experimental formulations designed to mimic endosomal membranes for improved intracellular processing. |
Protocol 2.2.1: Evaluating Phospholipid-Dependent Endosomal Escape This protocol uses live-cell imaging to quantify the endosomal escape efficiency of LNPs with different phospholipids [8].
- LNP Formulation: Prepare two LNP batches encapsulating Cy5-labeled mRNA, identical in composition except for the phospholipid: one with DSPC and the other with DOPE.
- Cell Seeding and Staining: Seed HeLa or HEK293T cells in glass-bottom imaging dishes. Allow to adhere for 24 hours.
- LNP Uptake: Incubate cells with the prepared LNPs (e.g., 0.5 µg mRNA/mL) for 2-4 hours in serum-free media.
- Endosome and Nucleus Labeling: Following incubation, add LysoTracker Green (75 nM) to stain acidic endosomes/lysosomes and Hoechst 33342 (5 µg/mL) to stain nuclei. Incubate for 30 minutes [8].
- Confocal Microscopy Imaging: Image cells using a high-resolution confocal microscope.
- Excitation/Emission: Cy5 (mRNA, red), LysoTracker Green (endosomes, green), Hoechst (nucleus, blue).
- Image Analysis: Quantify the degree of co-localization between the red (mRNA) and green (endosomes) signals using image analysis software (e.g., ImageJ). A lower co-localization coefficient in the DOPE sample indicates more efficient endosomal escape, as the mRNA has successfully exited the endosomal compartment [8].
Cholesterol: The Stabilizing Scaffold
Cholesterol is a crucial structural component in LNPs, constituting up to 40 mol% of the lipid composition [7]. It serves multiple vital functions: it integrates into the lipid bilayer to enhance stability and rigidity, reduces passive leakage of the cargo, and aids in cellular uptake, potentially by promoting membrane fusion [7] [3]. The molar percentage of cholesterol is a critical design parameter, as it directly influences LNP stability in circulation, protein corona formation, and ultimately, organ tropism [7].
Protocol 2.3.1: Optimizing Cholesterol Content for Liver-Targeted Delivery This protocol systematically evaluates how cholesterol content affects LNP physicochemical properties and in vivo liver expression [7].
- LNP Formulation: Formulate a series of LNPs using a fixed ionizable lipid (e.g., MC3 or SS-OP), phospholipid (DOPC or DSPC), and PEG-lipid (DMG-PEG2000). Systematically vary the cholesterol content (e.g., 10, 20, and 40 mol%) while adjusting the phospholipid proportion to maintain a total of 100%. Encapsulate FLuc mRNA in all formulations. For an example composition, see Table 3.
- Physicochemical Characterization: Characterize each LNP batch for:
- Size and PDI: Using Dynamic Light Scattering (DLS).
- Zeta Potential: Using Laser Doppler Micro-electrophoresis.
- Encapsulation Efficiency (%EE): Using a RiboGreen fluorescence assay [7].
- In Vivo Efficacy Study:
- Administer LNPs intramuscularly or intravenously to mice (e.g., ddY strain, n=5 per group) at a standardized mRNA dose.
- Measure luciferase expression via an in vivo imaging system (IVIS) at 6- and 24-hours post-injection.
- Quantify luminescence signal specifically in the liver and at the injection site (for intramuscular administration).
Table 3: Example LNP Compositions with Varying Cholesterol Content and Their Properties
| LNP ID | Cholesterol (mol%) | Phospholipid (mol%) | Ionizable Lipid (mol%) | PEG-lipid (mol%) | Size (nm) | PDI | EE (%) | In Vivo Liver Expression |
|---|---|---|---|---|---|---|---|---|
| LNP-C10 | 10 | 38.5 | 50 | 1.5 | ~100 | <0.25 | 88.6 ± 5.98 [7] | Low |
| LNP-C20 | 20 | 28.5 | 50 | 1.5 | ~100 | <0.25 | 89.0 ± 8.76 [7] | Medium |
| LNP-C40 | 40 | 8.5 | 50 | 1.5 | ~100 | <0.25 | 97.1 ± 0.93 [7] | High |
Data adapted from [7]. The results typically demonstrate that reducing cholesterol content leads to decreased protein expression in the liver after intramuscular or subcutaneous administration.
PEG-Lipids: The Stabilizing Shield
PEG-lipids, while typically comprising a small molar percentage (∼1.5%), are indispensable for creating therapeutically viable LNPs. They perform several key functions: they control and limit particle size during microfluidic formulation, prevent nanoparticle aggregation during storage and in circulation by steric stabilization, and reduce nonspecific uptake by the mononuclear phagocyte system, thereby extending circulation half-life [10] [3]. However, a "PEG dilemma" exists: excessive PEGylation can hinder cellular uptake and endosomal escape. Furthermore, PEG can induce immune responses such as the Accelerated Blood Clearance (ABC) phenomenon upon repeated dosing and Complement Activation-Related Pseudoallergy (CARPA) [10] [3].
Table 4: Guide to Selecting and Using PEG-Lipids
| Parameter | Impact on LNP Performance | Recommendation for Reprogramming mRNA LNPs |
|---|---|---|
| PEG Chain Length | Biphasic effect on immunogenicity; both very short and very long chains can induce anti-PEG antibodies [10]. | Use PEG2000, which is a standard and well-characterized length (e.g., DMG-PEG2000, ALC-0159) [3]. |
| Lipid Anchor | Determines the rate of PEG dissociation from the LNP. Faster-dissociating PEG (e.g., C14) allows for better cell interaction [10] [3]. | Use a short-chain anchor like DMG (C14) to allow for PEG shedding after delivery, facilitating cellular uptake and endosomal escape. |
| Molar Percentage | Higher percentages improve stability but can inhibit cellular uptake and transfection [10]. | Optimize between 1.0 and 2.0 mol%. Start with 1.5 mol% and adjust based on stability and potency assays. |
| ABC Phenomenon | Production of anti-PEG IgM after initial dose, causing rapid clearance of subsequent doses [10]. | For protocols requiring repeated LNP administration (e.g., multi-dose reprogramming), monitor for ABC or consider developing non-PEG alternatives. |
Protocol 2.4.1: Assessing the Impact of PEG-Lipid Percentage on LNP Potency and Stability
- LNP Formulation: Formulate a series of LNPs with a fixed core composition (ionizable lipid, phospholipid, cholesterol) but varying the PEG-lipid (e.g., DMG-PEG2000) content (e.g., 0.5, 1.5, and 3.0 mol%).
- Stability Testing:
- Size and PDI Monitoring: Measure the size and PDI of each formulation immediately after preparation (T=0) and after storage at 4°C for 1 week and 1 month. An increase in size or PDI indicates aggregation.
- Serum Stability: Incubate LNPs with 50% fetal bovine serum (FBS) at 37°C. Measure size and PDI over 4-6 hours.
- In Vitro Potency Assay: Transfert HepG2 or HEK293T cells with each LNP formulation (FLuc mRNA) and measure luminescence after 24 hours. Compare the relative light units (RLUs) to determine if higher PEG percentages inhibit transfection efficiency.
The Scientist's Toolkit: Essential Reagents for LNP Research
Table 5: Key Research Reagent Solutions for LNP Formulation and Testing
| Reagent / Material | Function / Description | Example Uses & Notes |
|---|---|---|
| Ionizable Lipids | Key functional component for RNA binding and endosomal escape. | MC3: Gold standard for siRNA. SM-102, ALC-0315: Used in COVID-19 mRNA vaccines. 5A2-SC8: Used in SORT LNP studies [9]. |
| Phospholipids | Structural "helper" lipids that form the LNP bilayer. | DSPC: For stable formulations. DOPE: For enhanced fusogenicity and endosomal escape [8] [9]. |
| Cholesterol | Natural sterol that stabilizes the LNP structure. | Sourced from suppliers like Sigma-Aldrich or Nacalai Tesque. Plant-derived cholesterol is also available for specific applications [7] [3]. |
| PEG-Lipids | Stabilizing agents that control size and prevent aggregation. | DMG-PEG2000: Commonly used, rapidly dissociating. ALC-0159: PEG-lipid used in the Comirnaty vaccine [3]. |
| Microfluidic Mixer | Instrument for precise, reproducible LNP self-assembly. | NanoAssemblr platforms are the industry standard for research-scale LNP formulation [7]. |
| mRNA Constructs | The therapeutic cargo. | CleanCap mRNA (e.g., from TriLink) with modified nucleosides (e.g., N1-methylpseudouridine) enhances stability and reduces immunogenicity [8] [4]. |
| Analytical Kits & Dyes | For characterizing LNPs and their biological activity. | RiboGreen Assay: For encapsulation efficiency [7]. LysoTracker & Hoechst: For live-cell imaging of uptake and endosomal escape [8]. |
The rational design of LNPs for reprogramming mRNA delivery hinges on a deep understanding of the four fundamental lipid components. As detailed in these application notes, the ionizable lipid drives efficacy, the phospholipid and cholesterol provide structural and stabilizing context, and the PEG-lipid ensures pharmaceutical stability. The future of LNP technology for reprogramming and other advanced therapies lies in further optimization and intellectual design. This includes developing new biodegradable ionizable lipids to improve safety profiles for repeated administration, which may be necessary for complete cellular reprogramming [5]. Furthermore, leveraging strategies like Selective Organ Targeting (SORT) by incorporating additional functional lipids can redirect LNPs from the liver to other tissues, expanding the potential of mRNA therapeutics beyond hepatic applications [9]. Finally, a thorough investigation of phospholipid chemistry and its influence on protein corona formation and organ tropism will be essential for creating next-generation LNPs tailored for specific clinical applications, such as the efficient generation of iPSCs for regenerative medicine [8] [9].
Application Note: Advanced LNP Platforms for Enhanced mRNA Delivery
This document provides a detailed technical overview of innovative lipid nanoparticle (LNP) strategies designed to overcome key biological barriers in the delivery of reprogramming mRNA. The content is structured to serve researchers and drug development professionals working on nucleic acid therapeutics, with a focus on practical methodologies and quantitative performance data.
The journey of an mRNA-loaded LNP from injection to protein expression is fraught with biological challenges. After administration, LNPs must protect the mRNA from enzymatic degradation, facilitate cellular uptake, and ensure endosomal escape to release the mRNA into the cytosol for translation. Current research focuses on engineering next-generation LNPs to enhance performance at each of these critical stages. Innovations in ionizable lipid design, mRNA core condensation, and surface functionalization are showing remarkable improvements in potency, targeting, and safety profiles, enabling more effective mRNA-based therapeutics and vaccines [11].
Quantitative Analysis of Next-Generation LNP Performance
The table below summarizes key quantitative findings from recent studies on advanced LNP systems, providing a benchmark for evaluating their potential in reprogramming mRNA research.
Table 1: Performance Metrics of Advanced LNP Formulations
| LNP Platform / Strategy | Key Performance Metric | Experimental Model | Reported Outcome | Significance for Reprogramming mRNA |
|---|---|---|---|---|
| Cyclic Ionizable Lipid (AMG1541) [12] | Vaccine dose reduction | Mouse (Flu vaccine) | Equivalent immune response at 1/100th the standard dose | Potential for reduced toxicity and cost in reprogramming factor delivery. |
| Metal-ion mRNA core (L@Mn-mRNA) [13] | mRNA loading capacity | In vitro & mouse models | ~2-fold increase vs. conventional LNPs; 2-fold higher cellular uptake | Higher mRNA payload could enhance reprogramming efficiency. |
| pHLIP Incorporation [14] | Endosomal escape & expression | Multiple cell lines & mice | 3 to 5-fold increase in mRNA expression | Addresses the critical bottleneck of cytosolic delivery for reprogramming factors. |
| Hybrisomes (MPE-functionalized) [15] | Cellular uptake & delivery | In vitro studies | Up to 15-fold higher uptake; 8-fold higher mRNA delivery | Could improve targeting and efficiency in hard-to-transfect primary cells. |
| Acuitas Next-Gen Lipids [16] | Potency increase | Preclinical models | Up to 4-fold higher potency in gene editing & vaccines | Suggests broader applicability for potent delivery of CRISPR/editing machinery. |
| Acuitas Novel Vaccine Lipids [16] | Dose-sparing effect | Preclinical models | Equivalent immunogenicity at a 5-fold lower dose | Enables lower, safer dosing regimens for in vivo reprogramming. |
Protocol: Formulation and Evaluation of High-Loading L@Mn-mRNA LNPs
This protocol details the creation of LNPs with a high-density mRNA core using a manganese ion (Mn2+)-mediated enrichment strategy, adapted from published research [13]. This method is suitable for various mRNAs and lipid compositions.
Materials and Reagents
- mRNA: Purified, IVT mRNA of interest (e.g., reprogramming factor mRNA).
- Metal Salt: Manganese chloride (MnCl₂) solution in nuclease-free water.
- Lipids: Ionizable lipid (e.g., DLin-MC3-DMA, ALC-315), cholesterol, helper phospholipid (e.g., DOPE), PEG-lipid.
- Buffers: Sodium acetate buffer (10 mM, pH 5.0), Tris-EDTA (TE) buffer.
- Equipment: Microfluidic mixer (e.g., NanoAssemblr), heating block, dynamic light scattering (DLS) instrument, transmission electron microscope (TEM).
Procedure
Synthesis of Mn-mRNA Core Nanoparticles:
- Dilute mRNA in nuclease-free water to a concentration of 0.1 mg/mL.
- Mix the mRNA solution with MnCl₂ solution at a molar ratio of 1 mRNA base to 5 Mn2+ ions. Note: A range of 8:1 to 2:1 (Mn2+:base) produces uniform nanoparticles [13].
- Incubate the mixture at 65°C for 5 minutes in a heating block.
- Allow the solution to cool to room temperature. The resulting complex is the Mn-mRNA nanoparticle (Mn-mRNA).
Lipid Coating via Microfluidic Mixing:
- Prepare an ethanolic lipid mixture containing the ionizable lipid, cholesterol, phospholipid, and PEG-lipid at desired molar ratios (e.g., 50:38.5:10:1.5).
- Load the Mn-mRNA nanoparticle solution (aqueous phase) and the ethanolic lipid mixture into separate syringes.
- Use a microfluidic mixer to combine the two streams at a fixed flow rate (e.g., 1:3 volumetric ratio, aqueous to ethanol) and a total flow rate of 12 mL/min.
- Collect the resulting L@Mn-mRNA formulation in a collection vial.
Purification and Characterization:
- Dialyze the formulated L@Mn-mRNA against a large volume of PBS (pH 7.4) for 4-6 hours at 4°C to remove ethanol and unencapsulated components.
- Use DLS to determine the particle size, polydispersity index (PDI), and zeta potential.
- Use a Quant-it RiboGreen RNA Assay to determine mRNA encapsulation efficiency. Expected outcome: >88% mRNA incorporation into the final LNP [13].
- Validate particle morphology using TEM.
Experimental Workflow Visualization
The following diagram illustrates the procedural workflow for creating L@Mn-mRNA nanoparticles.
Protocol: Functionalization of LNPs with Membrane Protein Extracts (Hybrisomes)
This protocol describes the creation of "hybrisomes," LNPs functionalized with cell-derived membrane proteins to significantly enhance cellular uptake and mRNA delivery efficiency, which is critical for targeting specific cell types in reprogramming [15].
Materials and Reagents
- Source Cells: Cultured cells from which membrane proteins will be extracted (e.g., specific primary cells relevant to reprogramming).
- Extraction Reagent: Mild detergent-based membrane protein extraction kit (e.g., Mem-PER Plus Kit).
- Lipids: Standard LNP lipid components.
- Buffers: Phosphate-buffered saline (PBS), protease inhibitor cocktail.
Procedure
Isolation of Membrane Protein Extracts (MPEs):
- Culture the source cells to 80-90% confluence.
- Harvest cells and wash twice with cold PBS.
- Following the manufacturer's instructions for the extraction kit, isolate the MPEs.
- Supplement the MPEs with a protease inhibitor cocktail to prevent degradation.
- Determine the protein concentration using a standard assay (e.g., BCA assay) and adjust to a working concentration.
Formulation of Hybrisomes:
- Prepare the standard ethanolic lipid mixture as for conventional LNPs.
- Instead of using plain buffer, resuspend the mRNA in the isolated MPE solution.
- Use a microfluidic mixer to combine the MPE/mRNA solution (aqueous phase) with the ethanolic lipid mixture.
- The MPEs incorporate into the lipid membrane during the self-assembly process.
- Purify the resulting hybrisomes via dialysis or tangential flow filtration against PBS.
Validation and Uptake Studies:
- Characterize hybrisomes using DLS and TEM. Note: Hybrisomes may exhibit unique bleb-like morphologies [15].
- To assess functionality, perform in vitro uptake studies. Incubate hybrisomes and standard LNPs (loaded with Cy5-mRNA) with target cells for 4-6 hours.
- Analyze cells using flow cytometry and confocal microscopy to quantify cellular uptake (Cy5 signal) and mRNA translation (eGFP expression if using eGFP mRNA). Expected outcome: Up to 15-fold higher cellular uptake and 8-fold higher mRNA delivery efficiency compared to standard LNPs [15].
The Scientist's Toolkit: Key Research Reagents
The table below catalogs essential reagents and their functions for developing advanced LNP systems for reprogramming mRNA research.
Table 2: Essential Reagents for Advanced LNP Research
| Research Reagent / Tool | Function & Application in LNP Development |
|---|---|
| Ionizable Lipids (e.g., ALC-315, novel cyclic lipids) | Key functional component of LNPs; protonated in acidic endosomes to promote membrane disruption and endosomal escape [12] [16]. |
| Polyethylene Glycol (PEG)-Lipids | Shields LNP surface, improves stability and circulation time; its rate of dissociation influences LNP biodistribution and cellular uptake [17]. |
| Manganese Chloride (MnCl₂) | Enriches mRNA into a dense core before lipid coating, dramatically increasing mRNA loading capacity in the final LNP formulation [13]. |
| pHLIP Peptide | Incorporated into LNPs to enhance endosomal escape; undergoes pH-dependent conformational change to disrupt endosomal membranes [14]. |
| Membrane Protein Extracts (MPEs) | Used to functionalize LNP surfaces ("hybrisomes") to enhance cell-specific targeting and dramatically improve cellular uptake [15]. |
| Galectin-9 Biosensor | A marker protein used in live-cell imaging to identify and study endosomal membrane damage and rupture, a key event for RNA release [18]. |
| BODIPY-labeled Ionizable Lipids | Fluorescently tagged lipids that allow for direct visualization of LNP trafficking and fate within cells using live-cell microscopy [18]. |
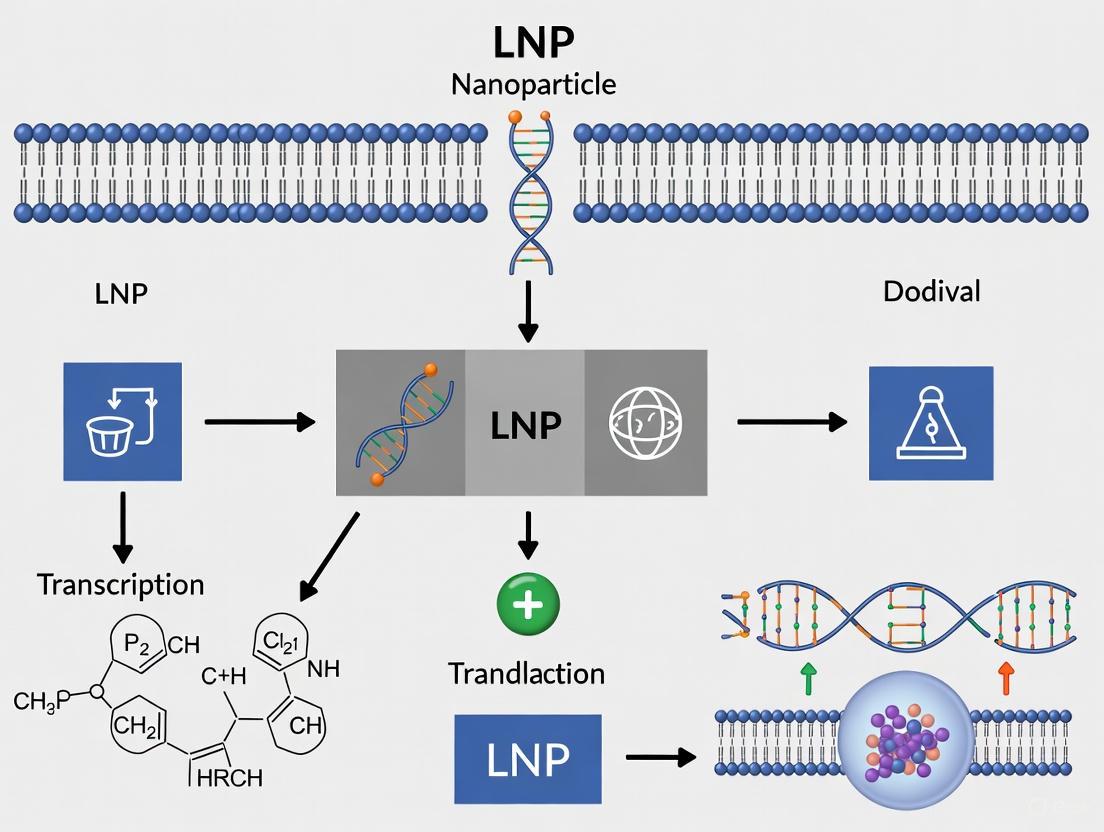
Visualizing the Cellular Journey and Key Barriers of LNPs
The following pathway diagram maps the intracellular pathway of an LNP and the primary barriers it must overcome for successful mRNA delivery, integrating recent mechanistic insights.
Advanced Strategy: Incorporating pHLIP for Enhanced Endosomal Escape
A major bottleneck in LNP-mediated mRNA delivery is the inefficient escape of mRNA from endosomes into the cytosol, with estimates of less than 5% of cargo successfully released [14]. Integrating the pH-low insertion peptide (pHLIP) into LNPs presents a direct strategy to overcome this barrier.
Materials and Reagents
- pHLIP Peptide: Synthesized pHLIP peptide.
- Standard LNP Components: Ionizable lipid, cholesterol, phospholipid, PEG-lipid, mRNA.
Procedure
Formulation of mRNA@LNP-pHLIP:
- The pHLIP peptide is included in the ethanolic lipid mixture during standard LNP formulation via microfluidic mixing.
- The peptide incorporates into the LNP membrane during self-assembly.
Mechanism of Action:
- After cellular uptake, the LNP is trafficked to acidifying endosomes.
- In the acidic environment (pH ~6.5-5.0), the pHLIP peptide undergoes a conformational change, transitioning from an unstructured state to an alpha-helix.
- This helical form inserts into the endosomal membrane, creating a pore or causing membrane disruption that facilitates the release of the mRNA payload into the cytosol.
Validation:
- In vitro: Transfect multiple cell lines and measure mRNA-encoded protein expression (e.g., luciferase or eGFP) via fluorescence or luminescence. Expected outcome: A 3 to 5-fold increase in protein expression compared to standard LNPs [14].
- In vivo: Administer mRNA@LNP-pHLIP intramuscularly and monitor for sustained and higher levels of protein expression in serum or tissues.
This application note provides a detailed technical overview of the key advantages of lipid nanoparticles (LNPs) over viral vectors for the delivery of reprogramming mRNA. Framed within the context of a broader thesis on LNP-mediated cell reprogramming, we delineate the core benefits of transient expression profiles, scalable manufacturing processes, and the elimination of insertional mutagenesis risks. The document includes structured quantitative data, detailed experimental protocols for assessing these advantages, and essential visual workflows to guide research and development efforts in this field.
The advent of mRNA-based cellular reprogramming necessitates delivery systems that are not only efficient but also safe and scalable. While viral vectors have been a historical mainstay, lipid nanoparticles (LNPs) have emerged as a superior non-viral alternative for in vivo and ex vivo reprogramming applications [19] [20]. LNPs are complex spherical vehicles typically composed of four lipid components: an ionizable lipid for mRNA complexation and endosomal escape, a phospholipid for structural support, cholesterol for membrane integrity and stability, and a PEGylated lipid to enhance nanoparticle stability and circulation time [21] [22]. The success of LNP-based mRNA vaccines has validated their clinical potential, highlighting key differentiators from viral systems [19] [20]. This document details the experimental frameworks for quantifying and leveraging the principal advantages of LNPs—controlled transient expression, unparalleled scalability, and a demonstrably safer profile by avoiding genomic integration—specifically for reprogramming mRNA research.
Core Advantages: A Comparative Analysis
The following table summarizes the fundamental advantages of LNPs over viral vectors, critical for designing reprogramming protocols.
Table 1: Core Advantages of LNP-mRNA over Viral Vectors
| Advantage | Mechanistic Basis | Consequence for Reprogramming Research |
|---|---|---|
| Transient Expression | mRNA operates in the cytoplasm without nuclear entry or genomic integration, leading to short-term, self-limiting protein expression [23]. | Prevents permanent genetic alteration; allows for precise control over reprogramming factor dosage and timing; reduces risks of oncogenic transformation from sustained expression of potent factors like Oct4, Sox2, Klf4, and c-Myc. |
| Scalable Manufacturing | Utilizes a modular, synthetic process with rapid, solvent-based microfluidics mixing. Components are chemically defined and do not require biosafety containment [20] [23]. | Enables rapid, cost-effective, and GMP-compliant production from pre-clinical to commercial scales; avoids the complex and time-intensive cell culture systems required for viral vector production. |
| Avoided Mutagenesis | Delivers mRNA cargo that remains episomal, completely avoiding the risk of insertional mutagenesis inherent to retroviral or lentiviral vectors [24] [23]. | Eliminates the risk of genotoxicity, including unintended gene disruption and oncogene activation, a critical safety consideration for therapeutic reprogramming applications. |
| Reduced Immunogenicity | Modern LNPs incorporate nucleoside-modified mRNA (e.g., pseudouridine) and purified components, significantly reducing innate immune activation compared to earlier formulations and many viral vectors [21]. | Minimizes inflammation and cell death at the transfection site, promoting a more favorable microenvironment for successful reprogramming and cell survival. |
To facilitate experimental design and expectation setting, the following table collates key quantitative metrics from seminal and recent studies on LNP-mRNA delivery.
Table 2: Key Quantitative Metrics for LNP-mRNA Delivery
| Parameter | Typical Range/Value | Context and Notes |
|---|---|---|
| Expression Onset | 2 - 8 hours post-transfection | Protein detection begins as ribosomes engage the delivered mRNA [23]. |
| Expression Duration | 24 - 96 hours | Dependent on mRNA design (UTR stability, cap structure) and cell type; extended with modified nucleotides [24] [23]. |
| Endosomal Escape Efficiency | ~2 - 3% | A critical bottleneck; only a small fraction of internalized LNPs successfully release their cargo into the cytosol [22]. |
| LNP Size (Diameter) | 70 - 150 nm | Optimized for cellular uptake; size is tunable via microfluidics parameters (flow rate ratio, total flow rate) [20] [22]. |
| Transfection Efficiency (in T cells) | Up to ~80% | Varies significantly with LNP formulation and cell activation state; critical for in vivo CAR-T cell generation [24]. |
| Cell Viability Post-Transfection | >80% | Can be significantly higher than electroporation, which may cause ~29% cell death [24]. |
Experimental Protocol: Evaluating Transient Expression Kinetics
Objective: To quantify the onset, peak, and duration of reprogramming factor expression delivered via LNP-mRNA in a target cell line (e.g., human fibroblasts).
Materials:
- Cells: Human dermal fibroblasts (HDFs), passage 3-5.
- LNP-mRNA: LNPs encapsulating mRNA encoding a reporter (e.g., eGFP) fused to a reprogramming factor (e.g., Oct4).
- Controls: Naked Oct4-eGFP mRNA, Viral vector (lentivirus) encoding Oct4-eGFP.
Method:
- Cell Seeding: Seed HDFs in a 24-well plate at a density of 5 x 10^4 cells/well and culture for 24 hours.
- Transfection: Treat cells with:
- Test Group: LNP-Oct4-eGFP mRNA (e.g., 100 ng mRNA/well).
- Control 1: Naked Oct4-eGFP mRNA (100 ng/well).
- Control 2: Lentivirus-Oct4-eGFP (MOI=5).
- Control 3: Untreated cells.
- Time-Course Analysis: At designated time points (e.g., 4, 8, 12, 24, 48, 72, 96 hours) post-transfection:
- Harvest Cells: Trypsinize and resuspend in flow cytometry buffer.
- Quantify Expression: Analyze eGFP fluorescence intensity using flow cytometry. Record the percentage of eGFP-positive cells and the mean fluorescence intensity (MFI) for each sample.
- Viability Assessment: Co-stain with a viability dye (e.g., propidium iodide) to assess cytotoxicity.
Data Analysis: Plot MFI versus time to visualize the kinetic profile of protein expression. Compare the peak MFI and the area under the curve (AUC) for LNP-mRNA versus controls. The transient nature of LNP-mRNA will be evidenced by a sharp rise and subsequent fall in MFI, contrasting with the persistent expression from the lentiviral control.
Diagram 1: Workflow for evaluating transient expression kinetics of LNP-mRNA.
Protocol for Scalable LNP Formulation and Characterization
Objective: To prepare, characterize, and test LNP-mRNA formulations for reprogramming applications using a reproducible microfluidics method.
Materials:
- Lipids: Ionizable lipid (e.g., DLin-MC3-DMA), Phospholipid (e.g., DSPC), Cholesterol, PEG-lipid (e.g., DMG-PEG2000).
- mRNA: Nucleoside-modified, HPLC-purified mRNA encoding the reprogramming factor of interest.
- Equipment: Microfluidic mixer (e.g., NanoAssemblr Ignite), PD-10 desalting columns, Dynamic Light Scattering (DLS) instrument.
Method:
- Lipid Solution Preparation: Dissolve the lipid mixture (ionizable lipid, DSPC, cholesterol, PEG-lipid at a molar ratio of 50:10:38.5:1.5) in ethanol to a final concentration of 10 mg/mL total lipid [20] [22].
- Aqueous mRNA Solution Preparation: Dilute mRNA in citrate buffer (10 mM, pH 4.0) to a concentration of 0.2 mg/mL.
- Microfluidic Mixing:
- Load the lipid and mRNA solutions into separate syringes.
- Set the total flow rate (TFR) to 12 mL/min and the flow rate ratio (FRR, aqueous:ethanol) to 3:1.
- Initiate mixing. The turbulent mixing within the microfluidic chip results in the instantaneous self-assembly of LNPs as the ethanol diffuses into the aqueous phase.
- Buffer Exchange and Purification: Dialyze or use tangential flow filtration (TFF) against PBS (pH 7.4) for at least 4 hours at 4°C to remove ethanol and buffer exchange. Alternatively, pass the LNP solution through a PD-10 desalting column equilibrated with PBS.
- Sterile Filtration: Filter the final LNP formulation through a 0.22 µm sterile filter.
Characterization:
- Particle Size and Polydispersity (PDI): Measure by Dynamic Light Scattering (DLS). Target: 80-120 nm with PDI < 0.2.
- Zeta Potential: Measure surface charge in PBS. Target: Near neutral to slightly negative charge.
- mRNA Encapsulation Efficiency: Use a Ribogreen assay. Compare fluorescence with and without Triton-X-100 detergent to distinguish encapsulated vs. free mRNA. Target: >90%.
- mRNA Integrity: Assess by agarose gel electrophoresis or capillary electrophoresis.
Diagram 2: Scalable LNP formulation workflow via microfluidics.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for LNP-mRNA Reprogramming Research
| Item | Function/Description | Example Product/Catalog Number |
|---|---|---|
| Ionizable Lipids | Core functional lipid for mRNA encapsulation and endosomal escape; pH-sensitive. | DLin-MC3-DMA, SM-102, ALC-0315 (custom synthesis) |
| Phospholipids | Provides structural integrity to the LNP bilayer. | 1,2-Distearoyl-sn-glycero-3-phosphocholine (DSPC) |
| PEGylated Lipids | Stabilizes LNPs, prevents aggregation, modulates PK/PD. | 1,2-Dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (DMG-PEG2000) |
| Microfluidic Mixer | Instrument for controlled, reproducible, and scalable LNP formation. | NanoAssemblr Ignite (Precision NanoSystems) |
| In vitro Transcription Kit | For high-yield, capped, and tailed mRNA synthesis. | mMessage mMachine T7 Transcription Kit (Thermo Fisher) |
| mRNA Modification | Incorporation of modified nucleosides (e.g., N1-methylpseudouridine) to reduce immunogenicity and enhance stability. | CleanCap Reagent AG (3' OMe) (Trilink BioTechnologies) |
| Flow Cytometer | For quantifying transfection efficiency and kinetic profiles of reporter constructs. | Attune NxT Flow Cytometer (Thermo Fisher) |
LNP-mRNA technology presents a compelling and robust platform for the next generation of cellular reprogramming therapies. Its defined advantages—controllable transient expression, a scalable and synthetic production pathway, and a foundational safety profile that precludes insertional mutagenesis—address critical limitations of viral vector systems. The protocols and data outlined herein provide a foundational framework for researchers to effectively develop, characterize, and utilize LNPs in reprogramming mRNA applications, accelerating the translation of this promising technology from bench to bedside.
Application Note: Immune Cell Reprogramming for Cancer Immunotherapy
Rationale and Scientific Foundation
The application of Lipid Nanoparticles (LNPs) to deliver mRNA for immune cell reprogramming represents a paradigm shift in cancer immunotherapy. This approach leverages the body's own immune system to recognize and eliminate tumor cells by providing genetic instructions for tumor-associated antigens (TAAs) or facilitating the engineering of immune cell receptors. The inherent immunostimulatory properties of mRNA-LNP formulations synergize with therapeutic goals by activating both innate and adaptive immune responses through multiple pathways, including toll-like receptor activation, type I interferon induction, and dendritic cell maturation [25].
Recent clinical breakthroughs have validated this approach, with the personalized mRNA cancer vaccine mRNA-4157 (combined with pembrolizumab) demonstrating a 44% reduction in recurrence risk compared to checkpoint inhibitor monotherapy in melanoma patients [25]. Similarly, BioNTech's BNT111 vaccine, an LNP-formulated mRNA encoding four tumor-associated antigens (NY-ESO-1, MAGE-A3, tyrosinase, and TPTE), has shown significant improvement in overall response rate in patients with anti-PD-(L)1 relapsed/refractory advanced melanoma [25]. These successes highlight the potential of mRNA-LNP platforms to effectively prime anti-tumor immune responses without requiring precise protein dosing, as immune activation benefits from variable and robust protein expression [25].
Key Experimental Data and Clinical Outcomes
Table 1: Clinical Outcomes of mRNA-LNP Cancer Immunotherapies
| Therapeutic Candidate | Target | Clinical Phase | Key Efficacy Outcomes | Reference |
|---|---|---|---|---|
| mRNA-4157 + pembrolizumab | Personalized neoantigens | Phase 2b | 44% reduction in recurrence risk vs pembrolizumab monotherapy in melanoma | [25] |
| BNT111 | NY-ESO-1, MAGE-A3, tyrosinase, TPTE | Phase 2 | Significant improvement in ORR in anti-PD-(L)1 relapsed/refractory advanced melanoma | [25] |
| BNT113 + pembrolizumab | HPV16+ head and neck cancer | Phase 2 | Exploratory efficacy in first-line treatment of advanced HNSCC | [26] |
| CVGBM | Newly diagnosed MGMT-unmethylated glioblastoma | Phase 1 | First in human study in surgically resected GBM | [26] |
Experimental Protocol: In Vitro Evaluation of mRNA-LNP for T-Cell Reprogramming
Objective: To evaluate the efficiency of mRNA-LNP formulations in reprogramming primary human T cells to express chimeric antigen receptors (CARs) or T-cell receptors (TCRs) for adoptive cell therapy applications.
Materials:
- Primary human T cells from leukapheresis product
- mRNA encoding CAR or TCR construct
- LNP formulations (ionizable lipid, phospholipid, cholesterol, PEG-lipid)
- Cell culture media (RPMI-1640 + 10% FBS + IL-2)
- Flow cytometry antibodies (CD3, CD4, CD8, CAR detection antibody)
- Electroporation system (for comparison studies)
Methodology:
T Cell Isolation and Activation:
- Isolate CD3+ T cells from PBMCs using negative selection magnetic beads.
- Activate T cells with CD3/CD28 activation beads for 24-48 hours.
- Maintain cells in complete media supplemented with 100 U/mL IL-2.
mRNA-LNP Formulation:
- Prepare LNPs using microfluidic mixing with the following standard composition:
- Ionizable lipid (35-50%)
- Phospholipid (10-15%)
- Cholesterol (25-40%)
- PEG-lipid (1-3%) [25]
- Encapsulate mRNA encoding CAR construct at nitrogen-to-phosphate ratio of 6:1.
- Characterize LNP size (Zetasizer), PDI, encapsulation efficiency (RiboGreen assay).
- Prepare LNPs using microfluidic mixing with the following standard composition:
T Cell Transfection:
- Wash activated T cells and resuspend in serum-free media at 10×10^6 cells/mL.
- Add mRNA-LNPs at optimized concentration (typically 100-200 ng mRNA/10^6 cells).
- Incubate for 4-6 hours at 37°C, then replace with complete media + IL-2.
- Include electroporation controls using same mRNA dose.
Analysis:
- Measure transfection efficiency at 24h by flow cytometry using CAR detection antibody.
- Assess T cell phenotype (memory subsets, exhaustion markers) at 48-72h.
- Evaluate in vitro cytotoxicity against antigen-positive target cells.
Critical Parameters:
- T cell activation status significantly impacts transfection efficiency
- mRNA design elements (5'/3' UTRs, nucleoside modifications) influence expression kinetics
- LNP composition affects intracellular delivery and potential immunogenicity
Application Note: Protein Replacement Therapy
Rationale and Scientific Foundation
mRNA-LNP technology enables protein replacement therapy by instructing patient cells to produce therapeutic proteins, addressing the root cause of various genetic and acquired diseases. This approach offers significant advantages over traditional protein therapeutics, including more natural post-translational modifications, adjustable dosing through mRNA administration, and avoidance of complex purification processes [26]. The amplification effect of mRNA technology is particularly beneficial, where a single mRNA molecule can direct the synthesis of 10^3-10^6 protein molecules through repeated ribosomal translation [25].
However, protein replacement therapy presents unique dosing precision challenges compared to vaccine applications. Current LNP delivery systems provide limited spatial and temporal control, with protein expression following predictable kinetics: rapid onset (2-6 hours), peak expression (24-48 hours), and exponential decline (7-14 days) [25]. This expression profile makes mRNA-LNPs particularly suitable for applications where transient protein production is therapeutic, but poses challenges for conditions requiring precise, sustained protein levels.
Clinical development has shown promising results across multiple disease areas. For cystic fibrosis, inhaled LUNAR-CFTR mRNA (ARCT-032) has demonstrated safety and tolerability in Phase 1 studies [26]. Similarly, mRNA therapy for Crigler-Najjar syndrome has shown correction of serum total bilirubin levels in mouse models [26], highlighting the potential for hepatic protein production.
Key Experimental Data
Table 2: mRNA-LNP Protein Replacement Therapies in Development
| Therapeutic Area | Target/Condition | Development Stage | Key Findings | Reference |
|---|---|---|---|---|
| Cystic Fibrosis | CFTR | Phase 1/2 | Inhaled LUNAR-CFTR mRNA safe and well-tolerated in human trials | [26] |
| Crigler-Najjar Syndrome | UGT1A1 | Preclinical | Correction of serum total bilirubin in mouse model | [26] |
| Metabolic Disorders | Various proteins | Clinical trials | Expression kinetics: onset 2-6h, peak 24-48h, decline 7-14 days | [25] |
| Ocular Diseases | Intravitreal delivery | Preclinical | Protein expression observed from 48h up to 2 weeks post-injection | [27] |
Experimental Protocol: Hepatic Protein Replacement via Systemic mRNA-LNP Administration
Objective: To achieve therapeutic protein production in hepatocytes through systemic administration of mRNA-LNP formulations.
Materials:
- mRNA encoding therapeutic protein (e.g., coagulation factor, metabolic enzyme)
- LNP formulations with hepatocyte tropism
- Animal model (e.g., C57BL/6 mice, Sprague-Dawley rats)
- ELISA kits for protein quantification
- Clinical chemistry analyzer for liver enzymes
- Tissue collection supplies (perfusion equipment, RNAlater)
Methodology:
mRNA Design and Production:
- Incorporate modified nucleosides (N1-methylpseudouridine) to reduce immunogenicity.
- Optimize 5' and 3' UTRs for enhanced translation and stability.
- Include poly(A) tail of optimal length (100-150 nucleotides).
- Produce mRNA via in vitro transcription and purify using HPLC/FPLC.
LNP Formulation for Hepatic Delivery:
- Utilize ionizable lipids with known hepatocyte tropism (e.g., DLin-MC3-DMA).
- Formulate LNPs using microfluidic mixing at 1:3 aqueous:organic flow rate ratio.
- Characterize particles for size (70-100 nm), PDI (<0.2), and encapsulation efficiency (>90%).
In Vivo Administration and Monitoring:
- Administer mRNA-LNPs via tail vein injection at doses ranging from 0.1-1.0 mg/kg.
- Collect blood samples at predetermined timepoints (4h, 24h, 48h, 72h, 7d, 14d).
- Monitor serum protein levels by ELISA.
- Assess liver function through ALT/AST measurements.
- Evaluate potential immune responses via cytokine profiling.
Tissue Analysis:
- At endpoint, perfuse liver and collect tissue samples.
- Analyze protein expression by immunohistochemistry.
- Assess mRNA biodistribution by qRT-PCR.
- Evaluate histopathology for potential toxicity.
Critical Parameters:
- LNP composition significantly influences hepatocyte delivery efficiency
- Nucleoside modifications are crucial for reducing innate immune activation
- Dosing regimen must balance protein expression duration with potential toxicity
Application Note: Gene Editing with CRISPR/Cas9 Systems
Rationale and Scientific Foundation
LNP-mediated delivery of CRISPR-Cas9 mRNA represents a revolutionary approach for therapeutic genome editing, enabling precise correction of disease-causing mutations. This combined technology platform allows for transient expression of the Cas9 nuclease, reducing off-target risks associated with prolonged nuclease activity while achieving durable therapeutic effects through permanent DNA modification [28] [29].
A significant advantage of LNP delivery over viral vectors is the potential for redosing, as demonstrated by recent clinical advances. Intellia Therapeutics reported that participants in a phase I trial for hereditary transthyretin amyloidosis (hATTR) safely received multiple doses of their LNP-delivered CRISPR therapy [28]. Similarly, the first personalized in vivo CRISPR treatment for CPS1 deficiency was successfully administered to an infant (patient KJ) via LNP delivery, with three doses safely administered to increase editing efficiency [28]. This redosing capability addresses a significant limitation of viral vector-based gene therapies.
Clinical success has been demonstrated across multiple genetic disorders. CTX310, an LNP-delivered CRISPR/Cas9 therapy targeting ANGPTL3 for severe dyslipidemia, has shown robust, dose-dependent reductions in circulating ANGPTL3 (mean reduction of -73% at highest dose) with corresponding significant reductions in triglycerides (-55%) and LDL cholesterol (-49%) [29]. The therapy was well-tolerated with no treatment-related serious adverse events [29].
Key Experimental Data
Table 3: Clinical Progress of LNP-Delivered CRISPR Therapeutics
| Therapeutic Candidate | Target/Gene | Condition | Clinical Outcomes | Reference |
|---|---|---|---|---|
| CTX310 | ANGPTL3 | Severe dyslipidemia | -73% ANGPTL3, -55% TG, -49% LDL reduction at 0.8 mg/kg dose | [29] |
| Intellia Program | TTR | hATTR amyloidosis | ~90% sustained reduction in TTR protein levels | [28] |
| Intellia Program | KLKB1 | Hereditary angioedema | 86% reduction in kallikrein, 8/11 participants attack-free | [28] |
| Personalized CRISPR | CPS1 | CPS1 deficiency | Safe administration of multiple LNP doses, symptom improvement | [28] |
Experimental Protocol: In Vivo Genome Editing in Hepatocytes
Objective: To achieve efficient genome editing in hepatocytes using LNP-formulated CRISPR-Cas9 mRNA and sgRNA.
Materials:
- mRNA encoding Cas9 (optimized codon usage, N1-methylpseudouridine)
- sgRNA targeting gene of interest (chemical modifications for stability)
- LNP components (ionizable lipid, DSPC, cholesterol, DMG-PEG2000)
- Animal model of genetic disease
- Next-generation sequencing platform
- Tissue processing equipment
Methodology:
CRISPR mRNA and sgRNA Preparation:
- Design mRNA with optimized UTRs and poly(A) tail for enhanced expression.
- Incorporate chemical modifications in sgRNA (2'-O-methyl, phosphorothioate) to enhance stability.
- Verify targeting efficiency and specificity using in vitro cleavage assays.
LNP Formulation for CRISPR Delivery:
- Co-encapsulate Cas9 mRNA and sgRNA in LNPs using microfluidic mixing.
- Utilize ionizable lipids with demonstrated hepatocyte tropism.
- Characterize LNP size, encapsulation efficiency, and RNA integrity.
In Vivo Delivery and Editing Assessment:
- Administer CRISPR-LNPs via systemic injection (tail vein in mice).
- Dose based on RNA content (typically 0.5-3 mg/kg total RNA).
- Monitor editing efficiency over time (days to weeks) to account for slow accumulation in non-dividing cells [30].
Editing Analysis:
- Collect liver tissue at various timepoints post-injection.
- Extract genomic DNA and assess editing efficiency by NGS.
- Evaluate potential off-target editing at predicted off-target sites.
- Measure functional outcomes (protein levels, biochemical correction).
Safety Assessment:
- Monitor liver enzymes (ALT, AST) and inflammatory markers.
- Assess histopathology for evidence of toxicity.
- Evaluate immune responses to Cas9 protein and LNPs.
Critical Parameters:
- Editing kinetics differ in non-dividing cells, with indels accumulating over weeks [30]
- LNP composition influences both delivery efficiency and immunogenicity
- sgRNA design is critical for both on-target efficiency and off-target minimization
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Research Reagents for LNP-mRNA Applications
| Reagent/Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| Ionizable Lipids | DLin-MC3-DMA, C12-200, S-Ac7-DOg | pH-dependent membrane fusion, endosomal escape | Primary determinant of delivery efficiency and tissue tropism |
| Helper Lipids | DSPC, DOPE | Structural integrity, membrane fusion | Influence LNP stability and cellular uptake |
| Polyethylene Glycol (PEG)-Lipids | DMG-PEG2000, DSG-PEG2000 | Steric stabilization, pharmacokinetics modulation | Affect circulation time and potential immune responses |
| Modified Nucleosides | N1-methylpseudouridine (m1Ψ), pseudouridine (Ψ) | Reduce immunogenicity, enhance translation efficiency | Critical for therapeutic application; impact protein yield |
| 5' Cap Analogs | CleanCap, ARCA Co-transcriptional capping | Enhance translation initiation, improve mRNA stability | Impact translational efficiency and intracellular recognition |
| In Vitro Transcription Kits | Custom optimized systems | mRNA production with high yield and purity | Reduce dsRNA contaminants that trigger immune responses |
| Characterization Tools | RiboGreen assay, dynamic light scattering | Measure encapsulation efficiency, particle size and distribution | Critical quality attributes for reproducible formulations |
Visualizing LNP-mRNA Workflows and Mechanisms
LNP-mRNA Experimental Workflow
Mechanism of LNP-mRNA Intracellular Delivery and Action
DNA Repair Pathways in CRISPR Editing
Advanced LNP Engineering and Emerging Applications in Cell Reprogramming
Within the field of lipid nanoparticle (LNP) delivery for reprogramming mRNA research, the ionizable lipid component serves as the critical determinant of both efficacy and safety. The rational design of these lipids, particularly through the incorporation of degradable cores and a deep understanding of structure-activity relationships (SAR), enables the development of next-generation vectors capable of efficient intracellular delivery while minimizing off-target toxicity. This document outlines the key principles, quantitative data, and experimental protocols essential for the design and evaluation of novel ionizable lipids, providing a framework for scientists engaged in the delivery of reprogramming mRNAs for cellular reprogramming and gene therapy applications.
Structure-Activity Relationships of Ionizable Lipids
The functional performance of an ionizable lipid—encompassing encapsulation, delivery efficiency, and toxicity—is governed by its chemical structure. The SAR can be systematically broken down into the influence of the lipid tail, the linker, and the amine head group [5] [3].
Table 1: Impact of Ionizable Lipid Structural Elements on Function
| Structural Element | Key Design Variations | Functional Impact on LNP Performance |
|---|---|---|
| Tail Saturation & Unsaturation | Linoleyl tails (2 cis double bonds) vs. saturated tails | Increased unsaturation enhances tendency to form non-bilayer phases, facilitating membrane disruption and endosomal escape [5]. |
| Tail Branching | Linear tails vs. branched tails (e.g., isodecyl) | Branched tails can boost mRNA expression >10-fold by promoting a cone-shaped structure and enhancing protonation at endosomal pH [5]. |
| Number of Tails | Two-tailed (e.g., MC3) vs. Multi-tailed (e.g., C12-200) | Multi-tailed lipids produce a more cone-shaped structure, potentially enhancing endosome-disrupting ability [5]. |
| Biodegradable Linkers | Ester bonds (primary vs. secondary), disulfide bonds | Introduces a trade-off: enhances biodegradability and safety but can reduce potency if hydrolysis is too rapid. Secondary esters and disulfide bonds offer a more favorable balance [5]. |
| Amine Head Group | Piperidine, Piperazine, aliphatic amines | The head group influences pKa and buffering capacity. Piperidine-based heads have been shown to limit reactive aldehyde generation, significantly improving LNP storage stability [31] [32]. |
The acid dissociation constant (pKa) of the ionizable lipid is a master variable in LNP performance. For effective in vivo hepatic delivery of mRNA, the optimal LNP pKa range has been expanded to approximately 6.2–7.4 [33]. The pKa dictates the surface charge of the LNP: neutral at physiological pH (reducing toxicity and non-specific interactions) but positively charged in the acidic endosome, which is crucial for interacting with anionic endosomal phospholipids [5] [6] [3]. This interaction facilitates the transition from a bilayer to an inverted hexagonal (HII) phase, destabilizing the endosomal membrane and enabling the cytosolic release of the mRNA cargo [5] [6].
Visualizing the Structure-Activity Relationship Framework
The following diagram summarizes the key structural considerations and their functional consequences in the rational design of ionizable lipids.
The Critical Role of Degradable Cores
For therapeutic applications requiring repeated dosing, such as sustained cellular reprogramming, the rapid and safe clearance of ionizable lipids post-delivery is paramount to prevent long-term accumulation and associated toxicity [5]. The primary strategy for introducing biodegradability involves the incorporation of hydrolysable bonds within the lipid structure.
Ester bonds are the most widely used degradable linkers. They are stable at physiological pH but are cleaved by intracellular esterases [5]. The design of these esters is critical:
- Primary Esters: Found in lipids like L319 (a biodegradable analog of MC3), they offer improved clearance but can sometimes lead to a loss in potency if hydrolysis occurs too rapidly [5].
- Secondary Esters: As seen in branched-tail lipids like FTT5, secondary esters exhibit a slower degradation rate, better balancing the trade-off between activity and degradability, and leading to higher transfection efficiency [5].
Disulfide bonds represent another powerful strategy. These bonds remain stable in the oxidative extracellular environment but are rapidly cleaved in the reductive cytoplasm (high glutathione concentrations) [5]. This mechanism facilitates timely payload release and lipid degradation. Ionizable lipids like 306-O12B have demonstrated efficient CRISPR/Cas9-mediated genome editing with negligible toxicity, outperforming the benchmark MC3 in some studies [5].
Quantitative Data and Design Parameters
Rational design is supported by quantitative data that links lipid structure to physicochemical properties and biological outcomes.
Table 2: Key Physicochemical Properties for Ionizable Lipid Evaluation
| Property | Target Range/Value | Analytical Technique | Significance for Reprogramming mRNA Delivery |
|---|---|---|---|
| pKa | 6.2 – 7.4 (hepatic delivery) [33] | TNS Assay, Potentiometric Titration | Determines charge-dependent endosomal escape and influences LNP stability and toxicity profile. |
| Size & PDI | 50 - 150 nm; PDI < 0.2 | Dynamic Light Scattering (DLS) | Affects cellular uptake, biodistribution, and packaging efficiency of large mRNA constructs. |
| Encapsulation Efficiency | > 90% | Ribogreen Assay | Ensures protection of mRNA from nucleases and maximizes cargo delivery to the target cell. |
| Buffering Capacity | Can help predict in vivo hepatic delivery [33] | Acid-Base Titration | May enhance endosomal escape through the "proton sponge" effect, buffering the endosomal pH. |
Machine learning (ML) is emerging as a powerful tool to navigate this complex design space. One study analyzing 213 LNPs with a random forest model identified phenol as a dominant substructure enhancing mRNA expression. The model also highlighted the importance of the N/P ratio (the ratio of amine groups in the lipid to phosphate groups in the mRNA) and the molar ratio of phospholipids like DSPC as critical compositional parameters [32].
Experimental Protocols
Protocol 1: Synthesis of a Biodegradable Ionizable Lipid (e.g., Ester-based)
This protocol outlines the synthesis of an ionizable lipid featuring a biodegradable ester bond, using a convergent strategy.
- Tail Synthesis: Synthesize or procure the desired hydrophobic tail precursor containing a carboxylic acid functional group (e.g., a branched alkyl chain). Purity via flash chromatography and confirm structure by NMR and mass spectrometry.
- Head-Linker Synthesis: Synthesize or procure the amine-containing head group precursor with a hydroxyl functional group (e.g., an N-methyl piperidine derivative).
- Esterification:
- In a round-bottom flask, combine the tail carboxylic acid (1.0 equiv), the head-group alcohol (1.2 equiv), and a catalytic amount of 4-dimethylaminopyridine (DMAP, 0.1 equiv) in anhydrous dichloromethane (DCM).
- Cool the mixture to 0°C under an inert atmosphere (N₂ or Ar).
- Add N,N'-dicyclohexylcarbodiimide (DCC, 1.1 equiv) or another suitable coupling agent in one portion.
- Allow the reaction to warm to room temperature and stir for 12-16 hours.
- Work-up and Purification:
- Filter the reaction mixture to remove the dicyclohexylurea precipitate.
- Wash the organic layer sequentially with 1M HCl, saturated NaHCO₃ solution, and brine.
- Dry the organic phase over anhydrous MgSO₄, filter, and concentrate under reduced pressure.
- Purify the crude product using a combination of normal-phase and reverse-phase flash chromatography.
- Characterization: Confirm the final product's identity and purity using techniques including ¹H/¹³C NMR spectroscopy and high-resolution mass spectrometry (HRMS). Assess chemical purity (>95%) by analytical HPLC.
Protocol 2: Formulation and Characterization of mRNA-LNPs
This protocol describes the preparation of mRNA-LNPs via microfluidic mixing and subsequent characterization.
- Lipid Stock Preparation:
- Prepare the lipid mixture by dissolving the ionizable lipid, phospholipid (e.g., DSPC or DOPE), cholesterol, and PEG-lipid (e.g., DMG-PEG2000) at a defined molar ratio (e.g., 50:10:38.5:1.5) in pure ethanol. The total lipid concentration is typically 10-20 mM.
- Aqueous Phase Preparation:
- Dilute the mRNA (e.g., reprogramming mRNA such as those encoding OCT4, SOX2, KLF4, c-MYC) in a citrate buffer (e.g., 25 mM, pH 4.0). The N/P ratio (moles of lipid amine N to mRNA phosphate P) should be calculated and used to determine the precise mRNA amount.
- Microfluidic Mixing:
- Load the lipid and aqueous phases into separate syringes on a microfluidic device.
- Set the flow rate ratio (aqueous:organic) to 3:1 and a total flow rate of 10-12 mL/min.
- Simultaneously push the solutions through the device, resulting in the instantaneous formation of mRNA-LNPs.
- Buffer Exchange and Dialysis:
- Collect the LNP suspension and dialyze against a large volume of PBS (pH 7.4) for 18-24 hours at 4°C using a dialysis membrane with an appropriate molecular weight cutoff (e.g., 100 kDa). Alternatively, use tangential flow filtration (TFF) for buffer exchange and concentration.
- Characterization:
- Size and PDI: Measure by Dynamic Light Scattering (DLS).
- Zeta Potential: Measure in a low-conductivity buffer.
- Encapsulation Efficiency: Use a Ribogreen fluorescence assay. Measure fluorescence with and without a detergent (e.g., Triton X-100) to disrupt the LNPs. EE% = (1 - (Free mRNA/Total mRNA)) * 100.
- pKa Determination: Use a fluorescent probe like TNS. Measure fluorescence of LNPs across a pH gradient (e.g., 3-10) and determine the pKa as the pH at the inflection point.
Protocol 3: Assessing In Vitro mRNA Delivery and Stability
This protocol evaluates the functional delivery of mRNA and the storage stability of the formulated LNPs.
- In Vitro Transfection:
- Seed target cells (e.g., HEK-293T, fibroblasts for reprogramming) in a 24-well plate.
- The next day, treat cells with mRNA-LNPs encoding a reporter gene (e.g., Firefly Luciferase, EGFP). Include appropriate controls (e.g., untreated cells, positive control like Lipofectamine MessengerMAX).
- Incubate for 24-48 hours.
- Analysis: For luciferase, lyse cells and measure luminescence. For EGFP, analyze by flow cytometry or fluorescence microscopy. Normalize data to total protein content.
- Storage Stability Study:
- Divide the LNP formulation into aliquots and store at different temperatures (e.g., -80°C, 4°C, 25°C).
- At predetermined time points (e.g., 1 week, 1 month, 3 months), analyze samples for:
- Physical Stability: Size and PDI by DLS.
- Chemical Stability: Purity of lipid components by HPLC.
- Functional Stability: In vitro transfection efficiency as described above, or an in vivo bioassay (e.g., serum hEPO levels after intravenous injection in mice if mRNA encodes human erythropoietin) [31].
- Aldehyde Impurity Assay:
- To assess the potential for mRNA adduct formation, incubate ionizable lipids with the fluorogenic reagent 4-hydrazino-7-nitro-2,1,3-benzoxadiazole (NBD-H).
- Measure the resulting fluorescence, which correlates with the amount of reactive aldehyde impurities generated by lipid degradation [31].
LNP Formulation and Characterization Workflow
The journey from lipid components to a characterized LNP formulation is a multi-step process, as visualized below.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Ionizable Lipid and LNP Research
| Reagent / Material | Function / Role | Examples & Notes |
|---|---|---|
| Ionizable Lipids | Core functional component; condenses mRNA, enables endosomal escape. | MC3 (Onpattro benchmark), SM-102 (Spikevax), ALC-0315 (Comirnaty). Novel designs: Piperidine-based lipids (e.g., CL15F series for stability), Biodegradable lipids (e.g., L319, 306Oi10) [5] [31] [3]. |
| Phospholipids | Helper lipids; provide structural integrity to LNP bilayer. | DSPC: Creates a tightly packed, stable bilayer. DOPE: Promotes hexagonal (HII) phase formation, facilitating endosomal membrane fusion [5] [3]. |
| Cholesterol | Helper lipid; enhances LNP stability and membrane integrity. | Plant-derived cholesterol is often used. Modulates membrane fluidity and prevents lipid component exchange [3]. |
| PEG-lipids | Stabilizing agent; controls particle size, reduces aggregation, and modulates pharmacokinetics. | DMG-PEG2000, ALC-0159. Critically, the PEG-lipid structure (chain length, linkage) influences the "PEG dilemma" of balancing stability versus cellular uptake [3]. |
| Microfluidic Device | Essential equipment for reproducible, scalable LNP formation. | Nanoassembler, Ignite; enables rapid mixing of lipid (ethanol) and mRNA (aqueous) phases [31]. |
| mRNA Constructs | Therapeutic cargo; the payload for encapsulation and delivery. | In vitro transcribed (IVT) mRNA, codon-optimized, with modified nucleosides (e.g., pseudouridine) to reduce immunogenicity. For reprogramming: mRNAs encoding factors like OCT4, SOX2 [34]. |
| Analytical Standards | For characterization of LNP physicochemical properties. | Late Nanosphere Size Standards (for DLS calibration), pH standards (for pKa calibration). |
APC-Mimetic LNPs for In Vivo CAR-T Cell Engineering
The field of adoptive cell therapy has been revolutionized by chimeric antigen receptor (CAR) T-cell immunotherapy, showing remarkable efficacy in treating hematological malignancies. However, the broader application of this technology is constrained by complex, costly, and time-consuming ex vivo manufacturing processes [35]. Current methods require T-cell isolation, activation, genetic modification, and expansion over 1-2 weeks in specialized GMP facilities, creating significant treatment delays and accessibility challenges [35] [36]. Additionally, prolonged ex vivo culture drives T-cells toward differentiated states with diminished persistence and antitumour potency [35].
Lipid nanoparticles (LNPs) have emerged as a promising non-viral delivery platform that could overcome these limitations. By encapsulating CAR-encoding nucleic acids and incorporating T-cell-specific targeting ligands, APC-mimetic LNPs enable in vivo CAR-T cell generation, potentially transforming cancer treatment paradigms [35] [36]. This approach bypasses ex vivo manufacturing entirely, allowing direct T-cell engineering within the patient's body through targeted LNP systems that simulate natural antigen-presenting cell (APC) functions [35].
The development of APC-mimetic LNPs aligns with broader research on LNP delivery of reprogramming mRNA, offering a versatile platform for precise genetic engineering of immune cells. This Application Note details the design principles, experimental protocols, and technical considerations for implementing APC-mimetic LNP technology for in vivo CAR-T cell engineering.
Background and Significance
CAR-T Cell Therapy: Current Limitations
CAR-T cell therapy has demonstrated remarkable success in treating aggressive lymphomas and acute lymphoblastic leukemia, with some cases showing complete and sustained remission [35]. However, several critical limitations hinder its broader application:
- Solid Tumor Challenges: CAR-T cells show poor efficacy against solid tumors due to insufficient trafficking to tumor sites, immunosuppressive microenvironments, and antigen escape variants [35]
- Safety Concerns: CAR-T activation can trigger severe toxicities including cytokine release syndrome (CRS) and neurotoxicity, which remain difficult to manage despite interventions like IL-6 blockade [35]
- Manufacturing Complexity: Current autologous approaches require individualized ex vivo engineering, making the process costly, time-consuming, and difficult to scale [35] [36]
- T-cell Exhaustion: Prolonged antigen exposure in the tumor microenvironment drives T-cells toward exhaustion and senescence, limiting long-term efficacy [35]
Rationale for APC-Mimetic LNPs
Natural T-cell activation requires multiple signals from professional antigen-presenting cells (APCs). Dendritic cells provide Signal 1 (antigen-specific TCR engagement via MHC) and Signal 2 (costimulatory CD80/CD86 binding to CD28) [35]. APC-mimetic LNPs replicate this coordinated activation by incorporating targeting and activating ligands in a single synthetic system.
The LNP platform offers distinct advantages over viral vectors, which dominate current CAR-T manufacturing:
- Avoidance of insertional mutagenesis risks associated with viral integration [35]
- Modular design allowing precise control over targeting and payload delivery
- Streamlined manufacturing processes compatible with large-scale production [36]
- Versatile payload capacity for mRNA, DNA, and combination cargoes [35] [36]
LNP Design and Formulation Strategies
Core LNP Composition and Payload Considerations
APC-mimetic LNPs for CAR-T engineering employ specialized formulations to achieve efficient nucleic acid delivery to T-cells. The table below summarizes key LNP composition variables and their functional impacts:
Table 1: LNP Composition Variables for T-Cell Engineering
| Component Category | Specific Examples | Functional Role | Performance Impact |
|---|---|---|---|
| Ionizable Lipids | nor-MC3 [37], PyCB IL [38], ALC-0315 [38] | Endosomal escape, complexation | Transfection efficiency, organ tropism |
| Nucleic Acid Payloads | CAR mRNA [35], Minicircle DNA [36], Transposase mRNA [36] | Genetic reprogramming | Expression kinetics, durability |
| Stabilizing Lipids | Egg sphingomyelin [37], DSPC [38] | Structural integrity, circulation time | Stability, biodistribution |
| Surface Ligands | Anti-CD7 nanobodies [36], Anti-CD3 scFv [36] | T-cell targeting, activation | Specificity, activation state |
Payload selection critically influences CAR expression kinetics and persistence. mRNA-based systems enable rapid but transient CAR expression (days to weeks), suitable for controlled therapeutic windows [35]. For sustained CAR expression, DNA-based systems incorporating transposase technology (e.g., SB100x) facilitate genomic integration, resulting in durable CAR-T cell persistence [36]. Recent advances demonstrate that minicircle DNA (mcDNA) combined with transposase mRNA in targeted LNPs can generate stable CAR-T cells with potent antitumor activity from a single administration [36].
Targeting Moieties for T-Cell Specificity
Precise T-cell targeting is essential for efficient in vivo engineering while minimizing off-target effects. The following targeting strategies have demonstrated efficacy:
- CD7-Targeted LNPs: Incorporate anti-CD7 nanobodies that leverage broad T-cell expression and internalization propensity [36]. These achieve efficient mRNA delivery without inducing T-cell activation, maintaining cells in a quiescent state [36]
- CD3-Targeted LNPs: Employ anti-CD3 single-chain variable fragments (scFvs) that provide both targeting and activation signals [36]. This approach mimics Signal 1 of natural T-cell activation [35]
- Dual-Targeted Systems: Combine CD7 and CD3 targeting ligands to optimize delivery across different T-cell activation states [36]. This strategy demonstrates superior transfection efficiency in both resting and activated T-cells compared to single-targeting approaches [36]
Table 2: Performance Comparison of Targeted LNP Formulations
| LNP Formulation | Transfection Efficiency (Resting T-cells) | Transfection Efficiency (Activated T-cells) | T-cell Activation | Key Applications |
|---|---|---|---|---|
| Untargeted LNPs | Minimal mcDNA delivery [36] | Modest mRNA delivery [36] | None | Baseline reference |
| tLNP-CD7 | Limited mcDNA delivery [36] | Efficient mRNA & mcDNA delivery [36] | No CD25 upregulation [36] | mRNA delivery without activation |
| tLNP-CD3 | Dose-dependent mcDNA delivery [36] | Moderate mcDNA delivery [36] | CD25 upregulation [36] | Combined targeting & activation |
| tLNP-CD7/CD3 | Highest efficiency in resting T-cells [36] | Superior transfection across payloads [36] | CD25 upregulation [36] | Optimal DNA delivery across activation states |
Advanced LNP Architectures for Enhanced Performance
Recent innovations in LNP design have addressed historical limitations in extrahepatic delivery:
- Liposomal LNPs: Systems with high proportions of bilayer-forming lipids (e.g., equimolar sphingomyelin and cholesterol at 4:1 bilayer-to-ionizable lipid ratio) exhibit liposomal morphology with extended circulation lifetimes and enhanced extrahepatic transfection [37]. These structures feature a solid core suspended in an aqueous interior surrounded by a lipid bilayer, improving stability and biodistribution [37]
- Zwitterionic Formulations: Three-component LNPs replacing cholesterol and PEGylated lipids with zwitterionic pyridine carboxybetaine (PyCB) ionizable lipids demonstrate reduced liver accumulation (∼70% lower) and increased spleen-specific mRNA translation (4.5-fold higher) [38]. This approach mitigates PEG immunogenicity and accelerates blood clearance issues associated with repeated administrations [38]
Experimental Protocols
LNP Formulation and Characterization Protocol
Objective: Prepare and characterize targeted LNPs for in vivo CAR-T cell engineering.
Materials:
- Ionizable lipid (e.g., nor-MC3, PyCB IL, ALC-0315)
- Structural lipids (e.g., ESM, DSPC, cholesterol)
- PEG-lipid (e.g., DMG-PEG)
- Targeting ligands (e.g., anti-CD7 nanobodies, anti-CD3 scFv)
- Nucleic acid payload (CAR mRNA, mcDNA with transposase)
- Microfluidic mixer (NanoAssemblr, Precision NanoSystems)
- Zetasizer (Malvern Panalytical)
Procedure:
Lipid Solution Preparation
- Dissolve ionizable lipid, structural lipid, cholesterol, and PEG-lipid in ethanol at molar ratios optimized for target application
- For bilayer-rich formulations: Use 20/40/40/1.5 molar ratio (ionizable lipid/ESM/cholesterol/PEG-lipid) for RB/I = 4 [37]
- For three-component systems: Replace cholesterol and PEG-lipid with zwitterionic PyCB IL [38]
Aqueous Phase Preparation
- Dilute nucleic acid payload in citrate buffer (pH 4.0) at appropriate concentration
- For DNA-based systems: Prepare minicircle DNA encoding CAR construct and transposase mRNA at optimized ratios [36]
LNP Formation
- Utilize microfluidic mixing with flow rate ratio 3:1 (aqueous:ethanol) at total flow rate 12 mL/min [37]
- Collect formulated LNPs in PBS buffer (pH 7.4)
Post-Formulation Functionalization
LNP Characterization
In Vitro T-Cell Transfection and Activation Assay
Objective: Evaluate targeted LNP-mediated CAR expression and functional consequences in primary human T-cells.
Materials:
- Human peripheral blood mononuclear cells (PBMCs) or isolated T-cells
- RPMI-1640 complete medium
- Flow cytometer with appropriate antibodies (CD3, CD25, CAR detection reagent)
- Cytokine release assay (IFN-γ, IL-2)
- Cytotoxicity assay components
Procedure:
T-Cell Preparation
- Isolate PBMCs from healthy donor blood via density gradient centrifugation
- Optionally isolate untouched T-cells using negative selection kit
- For resting T-cell experiments: Use freshly isolated cells without activation
- For activated T-cell controls: Stimulate with anti-CD3/CD28 beads 48-72 hours pre-transfection
LNP Transfection
- Dilute LNPs in serum-free medium to appropriate concentrations
- Incubate with T-cells (1-2×10^6 cells/mL) at various LNP concentrations
- Include untargeted LNP controls and non-transfected controls
- For time-course studies: Analyze cells at 24h (mRNA expression) and 4-7 days (DNA expression) [36]
Transfection Efficiency Analysis
- For reporter systems: Analyze fluorescent protein expression (eGFP, mCherry) by flow cytometry at 24h (mRNA) and 4 days (DNA) [36]
- For CAR expression: Stain with target antigen recombinant protein or anti-CAR antibody
- Evaluate T-cell subset specificity (CD4+ vs. CD8+) by surface marker staining
Activation Status Assessment
- Measure CD25 expression by flow cytometry 24-48h post-transfection [36]
- Analyze additional activation markers (CD69, CD71) as needed
- Compare activation profiles across LNP formulations (CD3-targeted vs. CD7-targeted)
Functional Characterization
- Measure antigen-specific cytokine production (IFN-γ, IL-2) upon co-culture with target cells
- Evaluate cytotoxic activity against antigen-positive target cells using real-time cytotoxicity assays
- Assess proliferation capacity through CFSE dilution or Ki67 staining
In Vivo CAR-T Cell Generation Protocol
Objective: Generate functional CAR-T cells in vivo through systemic administration of targeted LNPs.
Materials:
- Immunodeficient mice (NSG, NOG) humanized with PBMCs or CD34+ cells
- Tumor xenograft models (e.g., B-cell leukemia)
- IVIS imaging system for bioluminescence
- Flow cytometry with human-specific antibodies
Procedure:
Animal Model Preparation
- Establish humanized mouse models via PBMC or CD34+ cell transplantation [36]
- For efficacy studies: implant tumor cells (e.g., Nalm6-GL for B-ALL) and monitor engraftment
- Randomize animals based on tumor burden prior to LNP administration
LNP Administration
- Administer targeted LNPs via intravenous injection (dose: 0.5-2 mg/kg mRNA/DNA) [36]
- Include appropriate controls (PBS, untargeted LNPs, non-functional payload LNPs)
- For multiple dosing regimens: administer at 7-day intervals based on pharmacokinetic data
Monitoring and Analysis
- Track CAR-T cell generation in peripheral blood by flow cytometry
- Monitor tumor progression via bioluminescence imaging or caliper measurements
- Assess survival and clinical signs throughout study period
- At endpoint, analyze CAR-T cell infiltration in tumor, spleen, and bone marrow
Comprehensive Immune Profiling
- Evaluate CAR-T cell persistence in blood and tissues over time (≥4 weeks) [36]
- Characterize CAR-T cell differentiation status (naive, memory, effector subsets)
- Assess exhaustion markers (PD-1, TIM-3, LAG-3) in tumor-infiltrating CAR-T cells
- Measure host immune responses to LNP components or CAR construct
Signaling Pathways and Mechanisms
The molecular mechanisms of APC-mimetic LNP function involve coordinated signaling events that recapitulate natural T-cell activation while delivering genetic reprogramming cargo.
Diagram 1: APC-mimetic LNP Signaling Logic
The diagram above illustrates the core signaling logic of APC-mimetic LNPs, which deliver both activation signals and genetic reprogramming cargo to T-cells. This coordinated approach mimics natural APC function while enabling one-step CAR-T cell generation.
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of APC-mimetic LNP technology requires specialized reagents and materials. The following table details essential components for developing and evaluating these systems:
Table 3: Essential Research Reagents for APC-Mimetic LNP Development
| Reagent Category | Specific Examples | Function | Key Considerations |
|---|---|---|---|
| Ionizable Lipids | nor-MC3 [37], PyCB IL [38], DLin-MC3-DMA | Nucleic acid complexation, endosomal escape | pKa optimization, biodegradability, transfection efficiency |
| Targeting Ligands | Anti-CD7 nanobodies [36], Anti-CD3 scFv [36] | T-cell specificity, activation induction | Affinity, internalization capacity, orientation after conjugation |
| Nucleic Acid Payloads | CAR mRNA [35], Minicircle DNA [36], Transposase systems [36] | Genetic reprogramming | Expression kinetics, persistence, immunogenicity profile |
| Structural Lipids | Egg sphingomyelin [37], DSPC [38], Cholesterol | LNP stability, biodistribution | Phase behavior, bilayer formation, circulation half-life |
| Characterization Tools | Dynamic light scattering, RiboGreen assay, Cryo-TEM | LNP physicochemical characterization | Size distribution, encapsulation efficiency, morphology |
| Functional Assays | Flow cytometry, Cytokine ELISA, Cytotoxicity assays | CAR-T cell functional validation | Antigen specificity, activation status, tumor killing capacity |
Troubleshooting and Technical Considerations
Common Challenges and Solutions
- Low Transfection Efficiency in Resting T-cells: Implement dual CD3/CD7 targeting strategy and ensure proper T-cell activation via CD3 engagement [36]
- Rapid Clearance and Limited Extrahepatic Delivery: Utilize bilayer-rich LNP formulations (RB/I = 4) or zwitterionic three-component systems to extend circulation time [37] [38]
- Transient CAR Expression: Employ DNA-based systems with transposase technology (e.g., SB100x) for genomic integration and sustained expression [36]
- Off-target Transfection: Optimize targeting ligand density and validate specificity in mixed PBMC populations [36]
- LNP Stability Issues: Implement appropriate cryopreservation protocols and consider bilayer-stabilized formulations for long-term storage [37]
Critical Optimization Parameters
Successful APC-mimetic LNP development requires careful optimization of several key parameters:
- Targeting Ligand Density: Balance between specificity enhancement and potential steric hindrance of LNP function
- Payload Ratio: For DNA-based systems, optimize minicircle DNA to transposase mRNA ratio for efficient integration [36]
- LNP Size Distribution: Maintain narrow polydispersity (PDI <0.2) for consistent biodistribution and performance [36]
- Endosomal Escape Efficiency: Select ionizable lipids with appropriate pKa (typically 6.2-6.5) for efficient endosomal release
APC-mimetic LNPs represent a transformative approach to CAR-T cell engineering that addresses critical limitations of conventional manufacturing. By integrating T-cell targeting, activation, and genetic reprogramming in a single synthetic platform, these systems enable streamlined in vivo CAR-T cell generation with potential to significantly improve treatment accessibility and efficacy.
The protocols and design principles outlined in this Application Note provide a foundation for implementing this technology in research settings. As the field advances, continued optimization of LNP formulations, targeting strategies, and payload designs will further enhance the precision and potency of in vivo CAR-T cell engineering approaches.
The therapeutic potential of messenger RNA (mRNA) is fundamentally constrained by delivery challenges. While lipid nanoparticles (LNPs) have emerged as a dominant non-viral delivery platform, their inherent liver tropism has limited applications for extrahepatic diseases [39] [40]. This application note details cutting-edge methodologies for achieving organ-selective mRNA delivery to two critical non-hepatic targets: the lung and adipose tissue. Framed within a broader thesis on LNP-delivered reprogramming mRNA, this document provides actionable protocols and data for researchers aiming to engineer cell-specific therapeutics for metabolic and pulmonary diseases. We focus on two paradigm-shifting strategies: peptide-modified lipids for pulmonary targeting and machine learning-guided formulation screening for adipocyte-specific transfection.
Lung-Selective mRNA Delivery via Peptide-Ionizable Lipid Nanoparticles
Rationale and Principle
Overcoming the default hepatic accumulation of LNPs is a primary challenge in pulmonary gene therapy. A recent breakthrough involves the rational design of Peptide-Ionizable Lipids (PILs), which create a unique protein corona that mediates selective tissue uptake. Unlike conventional LNPs, which rely on ApoE-mediated hepatocyte targeting, PILs are engineered to selectively adsorb specific plasma proteins that direct them to extrahepatic tissues [41] [42]. The targeting specificity is exquisitely tunable, demonstrating single-amino-acid sensitivity, where minor sequence alterations can redirect LNPs from the lungs to the spleen or other organs [42].
Key Experimental Data and Formulation Optimization
The relationship between peptide structure and organ selectivity is quantifiable. The table below summarizes key findings from high-throughput screening of PIL libraries.
Table 1: Impact of Peptide Structure on Organ-Selective mRNA Delivery
| Structural Feature | Specific Example | Observed Targeting Profile | Key Quantitative Finding |
|---|---|---|---|
| Amino Acid Type | Poly-Lysine (K3) / Poly-Arginine (R3) | Lung | >90% of mRNA expression localized to lung [42] |
| Amino Acid Type | Glutamate (E) / Aspartate (D) / Proline (P) | Spleen | High spleen selectivity [42] |
| Peptide Chain Length | Di-arginine (2R) | Liver | Preferential liver accumulation [42] |
| Peptide Chain Length | Hexa-arginine (6R) | Lung | Optimal lung-targeting effectiveness [42] |
| Lipid Tail Chemistry | Saturated alkyl chains (a-tails) | Liver | Liver-targeting tendency [42] |
| Lipid Tail Chemistry | Amide-containing alkyl chains (aam-tails) | Spleen | Up to 86.0% spleen specificity [42] |
Detailed Protocol: PIL Synthesis and In Vivo Evaluation
Principle: This protocol describes a modular synthesis of PILs using Solid-Phase Support Synthesis (SPSS) and their subsequent assembly into LNPs for evaluating lung-targeted mRNA delivery [42].
Materials:
- Protected Amino Acids: Fmoc-protected natural and artificial ionizable amino acids (e.g., AIFAs).
- Lipid Tails: Various alkyl chains (e.g., 12-carbon saturated chain, hydroxylated chains).
- Structural Lipids: DOPE, Cholesterol, DMG-PEG2000.
- mRNA Cargo: Purified mRNA encoding a reporter protein (e.g., firefly luciferase).
Procedure:
- PIL Synthesis via SPSS:
- Begin with a Fmoc-protected AIFA linked to a solid support resin.
- Sequentially deprotect (using piperidine) and couple subsequent amino acids using standard Fmoc chemistry to build the desired peptide sequence.
- Following peptide assembly, conjugate the lipid tail to the N-terminus of the peptide.
- Cleave the final PIL from the resin using a suitable trifluoroacetic acid-based cocktail, followed by purification via HPLC.
LNP Formulation via Microfluidic Mixing:
- Prepare an ethanolic lipid phase containing the synthesized PIL, structural phospholipid (DOPE), cholesterol, and DMG-PEG2000 at a molar ratio of 15:20:25:2.
- Prepare an aqueous phase containing the mRNA payload in citrate buffer (pH 4.0).
- Use a microfluidic chaotic mixer to combine the two phases at a controlled flow rate (e.g., 1:3 aqueous-to-ethanol ratio) to form stable, mRNA-encapsulated LNPs.
- Dialyze the resulting LNP formulation against phosphate-buffered saline (PBS) to remove residual ethanol and adjust the pH.
In Vivo Administration and Analysis:
- Systemically administer the PIL-LNPs (e.g., via intravenous injection) into animal models (e.g., C57BL/6 mice).
- After 6-24 hours, euthanize the animals and harvest major organs (liver, spleen, lung, heart, etc.).
- Quantify mRNA delivery efficiency by measuring bioluminescent signal (for luciferase) or via qPCR for mRNA levels. Calculate the Targeting Specificity for each organ as the percentage of total signal across all organs.
Figure 1: Workflow for rational design and screening of peptide-ionizable lipids (PILs) for organ-selective mRNA delivery.
Adipocyte-Selective Transfection via Machine Learning-Guided LNP Screening
Rationale and Principle
Adipose tissue is a complex cellular ecosystem, with adipocytes constituting only 20-40% of the cell population amidst a stromal vascular fraction (SVF) containing preadipocytes, immune cells, and endothelial cells [43]. This cellular heterogeneity makes specific targeting via unique surface markers exceptionally difficult. To address this, a high-throughput screening (HTS) and machine learning (ML) approach was developed to identify LNP formulations that leverage physicochemical targeting for adipocyte-preferential transfection, independent of specific ligand-receptor interactions [44] [43].
Key Experimental Data and Formulation Optimization
A library of 649 LNP formulations was screened in vitro against adipocytes and macrophages to simultaneously optimize for both transfection efficiency and cell-type selectivity [43].
Table 2: Key Findings from High-Throughput Screening of Adipocyte-Selective LNPs
| Screening Parameter | Condition/Variable | Key Outcome and Quantitative Result |
|---|---|---|
| Ionizable Lipid | SM-102 | Used as the constant base ionizable lipid for all 649 formulations [43] |
| Helper Lipid Chemistry | Cationic (DDAB, DOTAP), Zwitterionic (DSPC, DOPE), Anionic (18BMP, 18PG) | Helper lipid chemistry was a critical driver of cell selectivity [43] |
| In Vitro vs. In Vivo Correlation | Top-performing formulations from in vitro screen | Cell-type selectivity (adipocyte vs. macrophage) was successfully recapitulated in vivo [43] |
| Benchmarking | FDA-approved Moderna Spikevax LNP | Used as a reference; selected novel formulations showed superior adipocyte selectivity in vivo [43] |
Detailed Protocol: HTS and ML Workflow for Adipocyte-Selective LNPs
Principle: This protocol uses a multi-step screening process in relevant cell types, followed by machine learning analysis to identify critical LNP compositional features that drive selective adipocyte transfection [43].
Materials:
- Cell Lines: Fully differentiated 3T3-L1 adipocytes and RAW 264.7 macrophages.
- Lipid Library: SM-102, Cholesterol, DMG-PEG-2000, and helper lipids (DDAB, DOTAP, DSPC, DOPE, 18BMP, 18PG).
- Reporter mRNA: mRNA encoding a fluorescent protein (e.g., eGFP) or luciferase.
- Microfluidic Device: For high-throughput LNP formulation.
Procedure:
- LNP Library Formulation:
- Prepare a combinatorial library of 649 LNP formulations using a microfluidic device. Keep the ionizable lipid (SM-102) constant while systematically varying the:
- Helper lipid type (cationic, zwitterionic, anionic).
- Concentrations of cholesterol and DMG-PEG-2000.
- Use the same batch of reporter mRNA as the payload for all formulations to ensure consistency.
- Prepare a combinatorial library of 649 LNP formulations using a microfluidic device. Keep the ionizable lipid (SM-102) constant while systematically varying the:
Dual-Objective In Vitro Screening:
- Apply the LNP library in parallel to plates containing fully differentiated 3T3-L1 adipocytes and RAW 264.7 macrophages.
- After a standard transfection period (e.g., 24-48 hours), measure transfection efficiency using flow cytometry (for fluorescence) or a plate reader (for luminescence).
- For each formulation, calculate a Selectivity Score (e.g., signal in adipocytes / signal in macrophages).
Machine Learning Analysis:
- Train a machine learning model (e.g., using SHAP analysis) using the LNP composition data (helper lipid type, cholesterol %, PEG-lipid %) as input features and the measured transfection efficiency/selectivity as the output [43].
- Use the model to identify which physicochemical features (e.g., helper lipid net charge) are most predictive of high adipocyte selectivity and high overall transfection.
In Vivo Validation:
- Select top candidate LNPs based on high efficiency and high selectivity scores from the in vitro screen.
- Administer these LNPs via direct intra-inguinal fat pad injection into a mouse model.
- Harvest the adipose tissue and analyze transfection levels specifically in the adipocyte fraction versus the SVF to confirm the persistence of selectivity observed in vitro.
Figure 2: High-throughput screening and machine learning workflow for identifying adipocyte-selective LNP formulations.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Organ-Selective LNP Research
| Reagent / Material | Function / Role | Specific Example(s) / Notes |
|---|---|---|
| Ionizable Lipids | Core component of LNPs; encapsulates mRNA and facilitates endosomal escape. | SM-102 (base for screening), custom Peptide-Ionizable Lipids (PILs) [43] [42] |
| Helper Lipids | Modulate LNP stability, fusogenicity, and cell selectivity. | DSPC, DOPE, DDAB, DOTAP, 18PG, 18BMP [43] |
| PEGylated Lipids | Shield LNP surface; control nanoparticle size and pharmacokinetics. | DMG-PEG2000, DSPE-PEG [43] [42] |
| Structural Lipids | Regulate LNP membrane integrity and fluidity. | Cholesterol (can be removed to reduce liver tropism) [45] |
| Reporter mRNAs | Enable rapid quantification of delivery efficiency and biodistribution. | Firefly Luciferase (FLuc), Green Fluorescent Protein (eGFP) [43] [42] |
| Microfluidic Mixer | Enables reproducible, high-throughput formation of monodisperse LNPs. | Used for both library synthesis and scaled production [43] [42] |
Machine Learning-Guided Formulation Screening for Cell-Type Specificity
The efficacy of lipid nanoparticles (LNPs) for delivering reprogramming mRNA is highly dependent on their composition, which dictates critical properties like cell-type specificity, transfection efficiency, and cytotoxicity. The parameter space for LNP formulation is enormous, encompassing variables such as ionizable lipid structure, helper lipid identity, and component ratios. Assuming at least 1000 different lipid combination choices and sampling only 10 conditions within each parameter (e.g., particle size, charge) can generate an intractable 10^9–10^10 combinations for experimental testing alone [46]. Machine learning (ML) has emerged as a powerful tool to navigate this complexity, enabling the in silico prediction of LNP performance and the rational identification of formulations with enhanced cell-type specificity for targeted mRNA delivery. This Application Note provides a detailed protocol for implementing ML-guided screening to discover LNPs for cell-specific reprogramming mRNA applications, using adipocyte-selective transfection as a primary case study [43].
Key Concepts and Rationale
The Formulation Challenge
LNPs are complex systems whose performance is influenced by the chemical nature and molar ratios of their components: an ionizable lipid, a helper lipid, cholesterol, and a polyethylene glycol (PEG)-lipid [47]. The ionizable lipid is particularly critical, as its chemical structure (head group, linker, and hydrocarbon tails) affects pKa, biodegradability, and endosomal escape efficiency [47]. Traditional discovery relies on high-throughput screening (HTS) of formulation libraries, which remains laborious, costly, and often fails to adequately explore the vast combinatorial space [46] [48].
The Machine Learning Advantage
ML models can be trained on data from HTS of LNP libraries to identify non-intuitive relationships between formulation parameters and functional outcomes. This data-driven approach can:
- Accelerate Discovery: ML models can rapidly predict the performance of unseen formulations, computationally prioritizing the most promising candidates for experimental validation [49] [48].
- Enhance Specificity: By screening libraries against multiple cell types in parallel, ML can elucidate compositional features that drive selectivity, enabling the design of LNPs that transfect target cells while minimizing off-target effects [43].
- Improve Efficiency: Pioneering work has demonstrated that ML models can achieve high accuracy (e.g., 98% test set accuracy) in predicting the transfection efficiency of LNP formulations [49].
Experimental Protocol: A Dual-Objective Screening Workflow
This protocol outlines the steps for screening an LNP library for cell-type-specific mRNA delivery, with integrated ML analysis. The workflow is summarized in the diagram below.
LNP Library Design and Synthesis
Objective: To create a diverse library of LNPs for screening. Materials:
- Ionizable lipid(s): SM-102 [43] or a novel library (e.g., AMG1541 [12]).
- Helper lipids: A panel with varied charges, e.g., cationic (DDAB, DOTAP), zwitterionic (DSPC, DOPE), anionic (18BMP, 18PG) [43].
- Structural lipids: Cholesterol, DMG-PEG-2000 [43].
- mRNA cargo: Reporter mRNA (e.g., Firefly Luciferase, GFP) for screening [43].
- Microfluidic device: For rapid and reproducible LNP formulation [46].
Procedure:
- Design Formulations: Create a library by systematically varying the molar ratios of components. A robust library may contain hundreds of formulations (e.g., 649 [43]). For instance, vary:
- Helper lipid type (cationic, zwitterionic, anionic).
- Cholesterol concentration (e.g., 20-50 mol%).
- PEG-lipid concentration (e.g., 1-5 mol%).
- Synthesize LNPs: Use a microfluidic device to mix an ethanol phase containing lipids with an aqueous phase containing mRNA at a defined flow rate ratio. This ensures rapid mixing and reproducible particle formation [46].
- Characterize LNPs: Post-formulation, characterize key physical properties:
- Particle Size and PDI: Use dynamic light scattering (DLS).
- Zeta Potential: Use electrophoretic light scattering.
- mRNA Encapsulation Efficiency: Quantify using a Ribogreen assay.
High-Throughput In Vitro Screening for Specificity
Objective: To evaluate the transfection efficiency and cell-type selectivity of the LNP library. Materials:
- Target Cell Type: e.g., Differentiated 3T3-L1 adipocytes [43].
- Off-Target Cell Type: e.g., Macrophages (e.g., J774A.1) as a model for adipose tissue stromal vascular fraction (SVF) [43].
- Cell culture media and reagents.
- Multi-well plate readers for high-throughput fluorescence or luminescence quantification.
Procedure:
- Cell Seeding and Differentiation: Seed and culture target and off-target cell types in appropriate multi-well plates (e.g., 96- or 384-well).
- LNP Transfection: Treat cells with LNP formulations from the library, normalizing to a constant mRNA dose across all conditions. Include a reference control (e.g., FDA-approved LNP like Spikevax [43]).
- Incubation and Analysis: Incubate for 24-48 hours, then quantify reporter protein expression (e.g., luminescence for Luciferase, fluorescence for GFP).
- Data Calculation: For each LNP in each cell type, calculate Transfection Efficiency (e.g., Relative Luminescence Units, RLU). Then, calculate a Selectivity Score, such as the log2 ratio of transfection efficiency in target cells versus off-target cells [43].
Machine Learning Model Training and Analysis
Objective: To build a predictive model that identifies formulation features driving efficiency and specificity. Materials:
- Computing environment: Python with scikit-learn, PyTorch, or TensorFlow libraries.
- Dataset: The curated dataset from Section 3.2.
Procedure:
- Data Curation: Assemble a dataset where each row is an LNP formulation, with features (e.g., mol% of each lipid, helper lipid identity, particle size) and labels (e.g., transfection efficiency in each cell line, selectivity score). This dataset should be split into training and test sets (e.g., 80/20 split).
- Model Training: Train a machine learning model, such as a Multilayer Perceptron (MLP) or other neural network/ensemble method, on the training set to predict the performance labels from the formulation features [49] [43].
- Model Validation: Evaluate the trained model's performance on the held-out test set using metrics like accuracy, mean squared error, or R² score.
- Feature Analysis: Use model interpretation tools (e.g., SHAP - SHapley Additive exPlanations) to identify which lipid components and physicochemical properties most strongly influence transfection efficiency and cell-type selectivity [43]. This provides actionable insights for the next design cycle.
In Vivo Validation of Top Candidates
Objective: To confirm the performance and selectivity of ML-prioritized LNP formulations in vivo. Materials:
- Animal model: e.g., C57BL/6 mice.
- mRNA cargo: Reporter mRNA (e.g., Luciferase) or therapeutic reprogramming mRNA.
- Route of administration: Relevant to the target tissue (e.g., direct intra-inguinal fat pad injection for adipocyte targeting [43], intramuscular injection).
- In vivo imaging system (IVIS) for bioluminescence/fluorescence imaging.
Procedure:
- Formulation Selection: Select the top 5-10 LNP candidates from the ML prediction based on high predicted efficiency and selectivity.
- Administration: Administer LNPs to animals via the chosen route.
- Biodistribution and Efficacy Analysis: At designated time points post-injection (e.g., 6, 24, 48 hours), image animals to quantify protein expression in the target organ and major off-target organs (e.g., liver, spleen).
- Tissue Analysis: Harvest tissues for further analysis (e.g., flow cytometry, immunohistochemistry) to confirm transfection in the specific target cell type (e.g., adipocytes) and quantify transfection in non-target cells within the tissue (e.g., SVF cells) [43].
Data Presentation and Analysis
Key Quantitative Findings from Case Studies
Table 1: Summary of ML-guided LNP screening outcomes from recent studies.
| Study Objective | Library Size | Key ML Model | Performance Outcome | Key Formulation Insight |
|---|---|---|---|---|
| Adipocyte Selectivity [43] | 649 formulations | Multilayer Perceptron | Identified formulations with >10-fold higher adipocyte vs. macrophage transfection. | Helper lipid chemistry (DOPE) and cholesterol content were critical drivers of selectivity. |
| mRNA Delivery Efficiency [49] | 622 LNPs (curated) | Multilayer Perceptron | 98% test set accuracy in predicting LNP transfection efficiency. | Model enabled in silico prioritization of high-performing candidates. |
| Protein Delivery for Gene Editing [48] | High-Throughput Screening | Machine Learning (unspecified) | Identified LNP formulations for efficient in vivo T cell gene editing (CCR5, PD-1). | Data-driven approach accelerated discovery for a challenging delivery application. |
Essential Research Reagent Solutions
Table 2: Key materials and reagents for implementing ML-guided LNP screening.
| Reagent / Solution | Function / Rationale | Example References |
|---|---|---|
| Ionizable Lipids (SM-102, novel) | Critical for mRNA encapsulation & endosomal escape; primary driver of LNP performance & pKa. | [43] [12] |
| Structurally Diverse Helper Lipids | Modulate LNP stability, fusogenicity, & cell-type specificity (e.g., DOPE for adipocytes). | [47] [43] |
| Cholesterol | Enhances structural integrity and stability of the LNP. | [47] [43] |
| DMG-PEG-2000 | Stabilizes LNP formation, reduces aggregation, & modulates pharmacokinetics. | [47] [43] |
| Reporter mRNA (Luc, GFP) | Enables high-throughput quantification of transfection efficiency and selectivity. | [43] [12] |
| Microfluidic Mixer | Enables rapid, reproducible, & scalable synthesis of LNP libraries. | [46] |
Discussion
The integration of machine learning with experimental screening represents a paradigm shift in LNP development. This workflow moves beyond a purely empirical approach to a more rational design process, powerfully illustrated by the successful identification of adipocyte-selective LNPs [43]. The critical success factors for this approach are high-quality experimental data for model training and the careful selection of formulation features. Future directions will involve incorporating more advanced biophysical descriptors of LNPs and in vivo biodistribution data into ML models to further enhance their predictive power for complex therapeutic applications like cell reprogramming.
Overcoming Delivery Hurdles: Strategies for Enhanced Efficiency and Safety
Inherited metabolic diseases (IMDs) and other monogenic disorders represent a significant area of unmet medical need, with a cumulative incidence of approximately 1 in 2000 live births [39]. Lipid nanoparticles (LNPs) have emerged as a revolutionary platform for delivering therapeutic messenger RNA (mRNA), demonstrating remarkable success during the COVID-19 pandemic and offering promising avenues for treating liver-directed IMDs [39] [50]. However, a fundamental limitation of conventional LNP systems is their inherent liver tropism, which restricts applications for diseases requiring extrahepatic delivery [51] [52].
This innate hepatic targeting primarily results from the standard LNP composition, particularly the presence of cholesterol and polyethylene glycol (PEG)-modified lipids [38]. Cholesterol promotes LNP adhesion to lipoproteins and subsequent uptake by hepatocytes, while PEGylated lipids, despite providing stability, can induce immunogenicity and accelerate blood clearance upon repeated administration [38]. For mRNA therapies targeting organs beyond the liver—such as the spleen for cancer vaccines or for autoimmune diseases—this hepatic accumulation represents a critical bottleneck, causing reduced efficacy at target sites and potential hepatotoxicity [53] [52].
Recent advances demonstrate that strategic reformulation of LNP components, particularly through cholesterol removal and replacement with innovative lipid designs, can successfully redirect biodistribution. This Application Note details specific protocols and data for overcoming innate liver tropism through composition reformulation, providing researchers with methodologies to enhance extrahepatic delivery for next-generation mRNA therapeutics.
Key Scientific Rationale and Quantitative Evidence
The Cholesterol Paradigm Shift
Conventional LNP formulations typically consist of four components: ionizable lipid, phospholipid, cholesterol, and PEG-lipid [51] [54]. Cholesterol comprises approximately 38.5 mol% of standard formulations such as BNT162b2 (Pfizer-BioNTech COVID-19 vaccine) and primarily functions to enhance LNP integrity and membrane fluidity by intercalating between lipids [54] [38]. However, cholesterol also promotes lipoprotein adhesion, thereby enhancing interactions with hepatocytes and ultimately limiting LNP trafficking to other organs [38].
Recent breakthrough research demonstrates that cholesterol is not essential for LNP functionality when replaced with appropriately designed zwitterionic lipids. A landmark study developed a three-component (ThrCo) LNP by replacing both cholesterol and PEGylated lipids in BNT162b2 LNPs with zwitterionic pyridine carboxybetaine (PyCB) ionizable lipids [38]. This strategic substitution achieved approximately 70% lower liver accumulation and a 4.5-fold increase in spleen-specific mRNA translation compared to the standard formulation [38].
Table 1: Quantitative Comparison of Standard vs. Cholesterol-Modified LNPs
| Formulation Parameter | Standard BNT162b2 LNP | ThrCo LNP (PyCB) | Change |
|---|---|---|---|
| Cholesterol Content | 38.5 mol% | 0 mol% | -100% |
| PEG-lipid Content | 1.5 mol% | 0 mol% | -100% |
| Liver Accumulation | Baseline | ~70% lower | ↓↓↓ |
| Spleen mRNA Translation | Baseline | 4.5-fold higher | ↑↑↑ |
| mRNA Encapsulation Efficiency | High (>90%) | 25.23% (initial) → Improved with optimization | Variable |
Alternative Ionizable Lipid Design Strategies
Beyond zwitterionic lipid replacements, modulation of ionizable lipid tail structures provides an alternative approach to reduce hepatic accumulation. Research has demonstrated that synthesizing ionizable lipids with varying tail lengths significantly impacts organ tropism [53]. In one systematic screening of 20 ionizable lipids with distinct tail structures, Lipid 7 emerged with a threefold higher mRNA expression efficiency at the injection site while simultaneously minimizing liver retention [53].
In HPV tumor models, Lipid 7 achieved comparable tumor suppression to SM-102-based LNP but demonstrated superior remodeling of the tumor microenvironment, increasing dendritic cell infiltration (12.1% vs. 5.1%) and elevating serum immune cytokines (TNF-α, IL-1β, etc., 1.2–1.8-fold higher) [53]. Critically, Lipid 7 reduced off-target mRNA accumulation in the heart, liver, spleen, lungs, and kidneys, mitigating hepatotoxicity risks associated with traditional LNPs [53].
Table 2: Efficacy and Safety Profile of Tail-Optimized Ionizable Lipid
| Evaluation Parameter | SM-102 LNP (Standard) | Lipid 7 LNP (Optimized) | Biological Impact |
|---|---|---|---|
| mRNA Expression at Injection Site | Baseline | 3-fold higher | Enhanced local efficacy |
| Tumor Suppression | Effective | Comparable | Maintained therapeutic effect |
| Dendritic Cell Infiltration | 5.1% | 12.1% | Improved immune activation |
| Natural Killer Cells | 0.5% | 1.1% | Enhanced immune response |
| Off-target Accumulation | Significant in liver | Reduced across all organs | Improved safety profile |
| Inflammatory Cytokines | Baseline | 1.2-1.8-fold higher | Potentiated immunity |
Experimental Protocols
Protocol 1: Formulation of Three-Component (ThrCo) LNPs with PyCB Lipids
Principle: Replace cholesterol and PEG-lipids with zwitterionic PyCB ionizable lipids to redirect biodistribution from liver to spleen [38].
Materials:
- Ionizable lipids: ALC-0315 and PyCB IL (synthesized as in [38])
- Phospholipid: 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC)
- mRNA: Purified, codon-optimized mRNA of interest
- Ethanol (absolute)
- Sodium acetate buffer (25 mM, pH 5.0)
- Tris-HCl buffer (pH 7.8)
- Microfluidic mixer (e.g., NanoAssemblr, Precision NanoSystems)
- Tangential Flow Filtration (TFF) system
Procedure:
- Lipid Solution Preparation: Dissolve ALC-0315, PyCB IL, and DSPC in ethanol at a molar ratio of 50:45:5 (ALC-0315:PyCB:DSPC). The optimal PyCB percentage may require empirical optimization between 40-50% [38].
- mRNA Solution Preparation: Dilute mRNA in 25 mM sodium acetate buffer (pH 5.0) to a concentration of 0.1 mg/mL.
- Microfluidic Mixing:
- Load the lipid solution (ethanol phase) and mRNA solution (aqueous phase) into separate syringes.
- Set the flow rate ratio to 1:3 (organic:aqueous) with a total flow rate of 12 mL/min.
- Mix using a microfluidic device to form LNPs.
- Buffer Exchange:
- Dilute the resulting LNP formulation with Tris-HCl buffer (pH 7.8).
- Perform buffer exchange using TFF with a 100 kDa molecular weight cutoff membrane.
- Concentrate the final formulation to the desired mRNA concentration.
- Quality Control:
Diagram 1: Three-Component LNP Formulation Workflow (Chars: 98)
Protocol 2: In Vivo Evaluation of Biodistribution and Efficacy
Principle: Quantify organ-specific LNP accumulation and protein expression to validate reduced liver tropism and enhanced target organ delivery [53] [38].
Materials:
- Luciferase-encoding mRNA (for biodistribution studies)
- Therapeutic mRNA of interest (for efficacy studies)
- Experimental animals (e.g., C57BL/6 mice, 6-8 weeks old)
- Near-infrared fluorescent dye (DiR) for LNP labeling
- In vivo imaging system (IVIS) or similar
- D-luciferin potassium salt
- Flow cytometer with appropriate antibodies
Procedure:
- LNP Administration:
- Inject mice intravenously via tail vein with DiR-labeled Luc-mRNA LNPs (5-10 μg mRNA per mouse).
- For therapeutic studies, administer LNP encapsulating therapeutic mRNA at optimized dose.
- Biodistribution Analysis:
- At predetermined time points (e.g., 6, 24 hours post-injection), image mice using IVIS system.
- Euthanize animals and harvest organs (heart, liver, spleen, lungs, kidneys).
- Image excised organs to quantify fluorescence intensity.
- Functional Efficacy Assessment:
- For Luc-mRNA LNPs, inject D-luciferin (150 mg/kg) intraperitoneally 6 and 24 hours post-LNP administration.
- Anesthetize mice and acquire bioluminescence images.
- Quantify signal intensity in regions of interest using appropriate software.
- Immune Cell Profiling:
- For splenic targeting studies, homogenize spleens and prepare single-cell suspensions.
- Stain cells with antibodies for dendritic cells (CD11c+), macrophages (F4/80+), and T cells (CD3+, CD4+, CD8+).
- Analyze by flow cytometry to identify cell populations transfected with mRNA.
- Statistical Analysis:
Diagram 2: In Vivo Biodistribution Evaluation Workflow (Chars: 99)
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Liver-Tropism Optimization Studies
| Reagent Category | Specific Examples | Function/Purpose | Protocol Relevance |
|---|---|---|---|
| Ionizable Lipids | ALC-0315, SM-102, PyCB IL, Lipid 7 | Core functional component for mRNA encapsulation and endosomal escape | Formulation optimization [53] [38] |
| Phospholipids | DSPC, DOPE, DSPE | Structural component for LNP bilayer formation | Standard and modified formulations [51] [54] |
| Stabilizing Lipids | DMG-PEG2k, Cholesterol (standard), PyCB (replacement) | Particle stability and biodistribution modulation | Replacement strategies [54] [38] |
| mRNA Constructs | Luciferase, eGFP, Therapeutic genes (e.g., HPV E6/E7) | Reporter genes for quantification and therapeutic effect assessment | Biodistribution and efficacy studies [53] [38] |
| Analytical Tools | Dynamic Light Scattering, Ribogreen Assay, IVIS Imaging System | Characterization of LNP properties and in vivo performance | Quality control and biodistribution [53] [38] |
| Cell Culture Models | 293T, CHO, DC2.4 cells | In vitro screening of LNP transfection efficiency | Preliminary formulation assessment [53] |
Strategic reformulation of LNP compositions, particularly through cholesterol removal and replacement with advanced ionizable lipids, represents a promising approach to overcome innate liver tropism. The protocols detailed herein provide researchers with validated methodologies to redirect LNP biodistribution toward extrahepatic targets, particularly the spleen, which is crucial for next-generation vaccines and immunotherapies.
Future directions in this field include the development of computational models like COMET (Composite Material Transformer) to accelerate LNP design [50], as well as continued innovation in zwitterionic and biodegradable ionizable lipids to further enhance targeting precision and safety profiles. As these technologies mature, they will unlock the full therapeutic potential of mRNA-LNP platforms for treating diverse diseases beyond hepatic disorders.
Optimizing Endosomal Escape with Novel Ionizable Lipid Chemistries
The efficacy of mRNA-based therapeutics and vaccines hinges entirely on the efficient cytosolic delivery of their genetic payload. Lipid nanoparticles (LNPs) are the leading non-viral delivery platform, with their clinical success demonstrated by COVID-19 mRNA vaccines. A critical bottleneck limiting their broader application, particularly for reprogramming mRNA research, is the inefficient escape of the mRNA from the endosomal compartment into the cytoplasm where translation occurs. Current estimates suggest that only 1-2% of the internalized nucleic acid payload successfully escapes endosomes [55]. The ionizable lipid component is the pivotal functional element of the LNP that orchestrates endosomal escape, making its optimization a primary focus for enhancing the potency of mRNA-LNP systems [56] [57] [19].
Ionizable lipids are engineered to be neutral at physiological pH (7.4) but become positively charged (protonated) in the acidic environment of endosomes (pH ~5.5-6.5). This protonation triggers a series of events: it promotes the fusion or destabilization of the endosomal membrane, often through the formation of non-bilayer hexagonal (HII) phases, facilitating the release of mRNA into the cytosol [57] [19]. The chemical structure of the ionizable lipid—encompassing the headgroup, linker, and tail chains—directly determines key properties such as pKa, biodegradability, and membrane-destabilizing capacity, thereby governing overall delivery efficiency and toxicity [57] [58]. This document provides detailed application notes and protocols for the design, formulation, and evaluation of novel ionizable lipids to maximize endosomal escape for advanced mRNA reprogramming applications.
Key Principles and Design Strategies for Ionizable Lipids
The evolution of ionizable lipids has progressed from simple cationic lipids to sophisticated, multi-functional structures. The design involves carefully balancing several structural domains to achieve optimal performance.
Critical Lipid Domains and Structure-Activity Relationships
- Ionizable Headgroup: The chemical nature of the headgroup dictates the pKa of the lipid, which is a crucial parameter. The ideal pKa for an ionizable lipid is typically between 6.0 and 6.5, ensuring neutrality in the bloodstream but sufficient protonation in early and late endosomes [58] [18]. Common headgroups include tertiary amines, which offer a tunable pKa. The headgroup's size and chemical properties also influence parameters like molecular packing and fusogenicity.
- Linker Chemistry: The linker connecting the headgroup to the hydrophobic tails impacts the stability, biodegradability, and function of the lipid. Ester-linked lipids are generally biodegradable, reducing long-term toxicity and facilitating metabolic clearance [19] [11]. In contrast, ether-linked lipids often exhibit higher metabolic stability and can demonstrate superior in vivo transfection efficiency in some contexts, as seen in early lipids like DOTMA [57] [19].
- Hydrophobic Tails: The structure, length, and degree of unsaturation of the hydrocarbon tails are critical for membrane fluidity and fusion. Tails with unsaturated chains (e.g., containing double bonds like linoleyl) introduce kinks, which prevent tight packing and promote the transition to the hexagonal HII phase conducive to endosomal escape [57]. Symmetrical tails with identical chains are often preferred for optimal transfection activity [57].
Table 1: Key Domains in Ionizable Lipid Design and Their Functional Impact
| Lipid Domain | Design Options | Impact on LNP Function | Representative Examples |
|---|---|---|---|
| Headgroup | Tertiary amines, cyclic amines, piperazine | Determines pKa and protonation capacity; influences charge density and fusogenicity. | DLin-MC3-DMA (MC3), ALC-0315 |
| Linker | Ester, ether, ketal, degradable amide | Controls biodegradability, metabolic clearance, and chemical stability. | L319 (ester), DOTMA (ether) |
| Hydrophobic Tails | Saturated (e.g., C18), Unsaturated (e.g., DLin), Branch-chain | Governs membrane fluidity, fusion efficiency, and propensity to form hexagonal HII phases. | DLin in MC3, hexyldecanoate in ALC-0315 |
Quantitative Analysis of Ionizable Lipid Properties
The following table summarizes critical quality attributes (CQAs) for a selection of clinically relevant and novel ionizable lipids. These parameters serve as benchmarks for designing new lipids targeted at enhanced endosomal escape.
Table 2: Quantitative Properties of Selected Ionizable Lipids for Performance Benchmarking
| Ionizable Lipid | Reported pKa | Key Structural Features | Primary Application & Notes |
|---|---|---|---|
| DLin-MC3-DMA (MC3) | ~6.44 [18] | Linoleyl tails, dimethylaminopropane headgroup, degradable ester linker | First FDA-approved siRNA drug (Onpattro); benchmark for liver delivery. |
| ALC-0315 | Data for precise pKa in sources | Hexyldecanoate branched tails, dimethylamine headgroup, ester linkers. | Key component in COVID-19 vaccine (BNT162b2); robust in vivo efficacy. |
| SM-102 | Data for precise pKa in sources | Proprietary structure with synthetic tails and amine headgroup. | Key component in COVID-19 vaccine (mRNA-1273); high delivery efficiency. |
| L319 | ~6.0-6.5 (estimated) | Ester-modified MC3 analog; designed for enhanced biodegradability. | Shows improved tolerability and rapid clearance vs. MC3 [19]. |
| DLin-KC2-DMA | Data for precise pKa in sources | Optimized dioxolane linker from DLin-DMA. | Demonstrates the impact of linker optimization on siRNA potency [19]. |
Experimental Protocol: Formulation and In Vitro Evaluation of LNPs
This section provides a detailed methodology for formulating mRNA-LNPs using novel ionizable lipids and assessing their endosomal escape efficiency through functional and imaging-based assays.
Protocol: Microfluidic Formulation of mRNA-LNPs
Objective: To reproducibly prepare mRNA-encapsulating LNPs with high encapsulation efficiency and a narrow particle size distribution. Principle: Lipids dissolved in an organic solvent are rapidly mixed with an aqueous mRNA solution in a microfluidic device. The change in polarity causes lipid self-assembly into nanoparticles, encapsulating the mRNA [19] [11].
Materials:
- Ionizable Lipid (novel compound under investigation)
- Helper Lipid: DSPC (Distearoylphosphatidylcholine) or DOPE (Dioleoylphosphatidylethanolamine)
- Cholesterol: (Stability and rigidity of LNP)
- PEG-lipid: DMG-PEG2000 or ALC-0159 (prevents aggregation, modulates pharmacokinetics)
- mRNA: Clean, modified mRNA encoding a reporter gene (e.g., EGFP, Luciferase) or reprogramming factor.
- Ethanol (absolute, molecular biology grade)
- Sodium Acetate Buffer (10 mM, pH 4.0)
- PBS (1X, pH 7.4)
- Dialysis Membranes (MWCO 100 kDa) or Tangential Flow Filtration (TFF) system
- Microfluidic Device (e.g., NanoAssemblr, Ignite, or homemade staggered herringbone mixer)
Workflow:
Procedure:
- Prepare Lipid Mixture:
- Dissolve the ionizable lipid, helper lipid (DSPC or DOPE), cholesterol, and PEG-lipid in ethanol at a molar ratio of 50:10:38.5:1.5 (a common starting point). The total lipid concentration is typically 10-20 mM.
- Vortex and gently warm the solution to ensure complete dissolution.
Prepare mRNA Solution:
- Dilute the mRNA in sodium acetate buffer (10 mM, pH 4.0) to a final concentration of 0.1-0.2 mg/mL. The acidic pH helps maintain the ionizable lipid in a protonated state during formulation, improving encapsulation.
Microfluidic Mixing:
- Set the flow rate ratio (aqueous:organic) to 3:1. A total combined flow rate of 12 mL/min is a standard starting parameter.
- Load the lipid-ethanol solution and the mRNA-aqueous solution into separate syringes.
- Initiate simultaneous pumping through the microfluidic device. The instantaneous mixing generates a milky solution, indicating LNP formation.
Buffer Exchange and Dialysis:
- Collect the LNP solution and immediately dialyze against a large volume of 1X PBS (pH 7.4) for at least 4 hours at 4°C to remove ethanol and adjust the pH. Alternatively, use TFF for faster processing and buffer exchange.
- After dialysis, filter the LNP solution through a 0.22 µm or 0.45 µm sterile filter.
Characterization and Quality Control:
- Particle Size and PDI: Measure by Dynamic Light Scattering (DLS). Aim for a diameter of 70-100 nm with a PDI < 0.2.
- Encapsulation Efficiency: Use a Ribogreen assay. Quantify total mRNA, then lyse LNPs with 1% Triton X-100 to measure encapsulated mRNA. >90% encapsulation is desirable.
- Zeta Potential: Measure in 1X PBS. LNPs should have a near-neutral surface charge for reduced non-specific interactions.
Protocol: Assessing Endosomal Escape Using Live-Cell Microscopy
Objective: To visually confirm and quantify the endosomal escape of mRNA-LNPs in live cells. Principle: This assay uses galectin proteins as sensitive biomarkers for endosomal membrane damage. When LNPs disrupt the endosomal membrane, galectins (e.g., Galectin-9, Galectin-8) rapidly bind to exposed glycans on the inner leaflet, serving as a fluorescent proxy for escape-competent events [18].
Materials:
- Cells for transfection (e.g., HEK293, HeLa, or target primary cells)
- LNP formulations with Cy5-labeled mRNA
- Plasmid or cell line expressing Galectin-9-mGFP or Galectin-8-mGFP
- Confocal or super-resolution live-cell microscope
- Glass-bottom culture dishes
- Complete cell culture medium
Workflow:
Procedure:
- Cell Preparation:
- Seed cells stably expressing Galectin-9-mGFP into glass-bottom dishes 24 hours before imaging to achieve 60-70% confluency.
- Alternatively, transiently transfect cells with the Galectin-9-mGFP plasmid 48 hours prior to the experiment.
LNP Treatment and Imaging:
- Replace the medium with fresh, pre-warmed medium.
- Add the Cy5-mRNA LNP formulation at a predetermined concentration (e.g., 0.5-1.0 µg/mL mRNA).
- Immediately transfer the dish to a live-cell microscope chamber maintained at 37°C and 5% CO₂.
- Begin time-lapse imaging, acquiring both GFP (galectin) and Cy5 (mRNA) channels every 5-10 minutes for 4-8 hours.
Image and Data Analysis:
- Identify Damaged Endosomes: Manually or automatically threshold and identify foci where Galectin-9-mGFP signal appears de novo.
- Quantify "Hit Rate": For each galectin-positive endosome, determine if it contains a detectable Cy5-mRNA signal. The "hit rate" is the percentage of galectin-damaged endosomes that contain mRNA cargo. Note that this rate can be low (e.g., ~70% for siRNA, ~20% for mRNA), revealing a significant barrier [18].
- Track Cargo Release: In endosomes that are both galectin- and mRNA-positive, monitor the subsequent decrease in Cy5 signal intensity, which indicates mRNA release into the cytosol.
Table 3: Key Research Reagent Solutions for LNP Development
| Reagent / Material | Function / Application | Example Sources / Identifiers |
|---|---|---|
| Ionizable Lipids (MC3, ALC-0315) | Benchmarking and control formulations for comparative studies. | Available from specialty chemical suppliers (e.g., MedKoo, Avanti). |
| Helper Lipids (DOPE, DSPC) | Promote hexagonal phase formation and membrane fusion (DOPE) or provide bilayer structure (DSPC). | Avanti Polar Lipids (850725, 850365). |
| PEG-Lipids (DMG-PEG2000) | Provides a stealth layer, controls particle size, and improves stability. | Avanti Polar Lipids (880151). |
| Microfluidic Formulator | Enables reproducible, scalable LNP production with high encapsulation efficiency. | NanoAssemblr (Precision NanoSystems), Ignite (Precision NanoSystems). |
| Galectin-9-mGFP Plasmid | Critical biosensor for visualizing endosomal membrane damage in live cells. | Addgene (various constructs). |
| Ribogreen Assay Kit | Quantifies mRNA encapsulation efficiency in formulated LNPs. | Thermo Fisher Scientific (R11490). |
| Dynamic Light Scattering (DLS) | Measures LNP particle size, polydispersity (PDI), and zeta potential. | Instruments from Malvern Panalytical, Horiba. |
Advancing reprogramming mRNA research demands LNPs that surpass the efficiency of current benchmarks. A rational design approach for novel ionizable lipids, focused on fine-tuning pKa, enhancing biodegradability, and optimizing tail unsaturation, is paramount to overcoming the critical barrier of endosomal escape. The protocols detailed herein for LNP formulation, characterization, and functional assessment using advanced live-cell imaging provide a robust framework for screening and validating next-generation ionizable lipids. By systematically applying these strategies, researchers can develop more potent and specific LNP delivery systems, unlocking the full potential of mRNA-based cellular reprogramming and therapeutics.
Mitigating Immunogenicity and Inflammatory Responses
The clinical success of lipid nanoparticles (LNP) in delivering mRNA therapeutics has been tempered by significant immunogenicity and inflammatory responses, which can impact both safety and efficacy. These responses originate from multiple components of the LNP-mRNA platform, including the mRNA molecule itself and the ionizable lipid component of the delivery system [59] [60]. While this inherent immunostimulation can be beneficial for vaccine applications, it presents substantial challenges for therapeutic applications requiring repeated administration or precise dosing control, such as protein replacement therapies or regenerative medicine [61] [62]. This application note provides detailed methodologies for quantifying, understanding, and mitigating these immune responses to advance the development of LNP-based reprogramming mRNA therapeutics.
Mechanisms of Immunogenicity
mRNA-Mediated Immune Activation
Exogenous mRNA can trigger innate immune recognition through multiple pathways:
- Pathogen recognition receptors: mRNA is recognized by Toll-like receptors (TLR3, TLR7, TLR8), RIG-I, and MDA-5 in a sequence- and structure-dependent manner [59]
- Immunogenicity mitigation: Nucleoside modifications (e.g., pseudouridine, N1-methylpseudouridine) reduce innate immune detection and activation of type 1 interferons [59] [62]
- Translation impact: Unmodified mRNA causes significantly greater global translational repression (~58% inhibition) compared to modified mRNA, which shows 40-46% higher translation levels in comparative studies [59]
LNP Component-Mediated Inflammation
The ionizable lipid component of LNPs represents a major source of inflammatory responses:
- TLR4 activation: Ionizable lipids in LNPs signal through TLR4 to activate both NF-κB and IRF transcription factors, triggering inflammatory cytokine production [60]
- Kinetics: NF-κB activation peaks at 48-72 hours post-exposure, demonstrating delayed kinetics compared to traditional TLR agonists [60]
- Component specificity: LNPs lacking ionizable lipids show complete loss of NF-κB and IRF activation, confirming the primary role of this component [60]
- Inflammatory cytokines: LNP exposure triggers release of proinflammatory cytokines including IL-6, TNF-α, IFN-γ, and IL-1 [63]
Table 1: Quantitative Comparison of Immune Activation by Different Ionizable Lipids
| Ionizable Lipid | NF-κB Activation (fold-change) | IRF Activation (fold-change) | Key Characteristics |
|---|---|---|---|
| LNP-ALC0315 | 4-fold at 48h | 3-fold at 48h | BNT162b2 formulation |
| LNP-SM102 | Similar to ALC0315 | Significantly greater than ALC0315 | mRNA-1273 formulation |
| LNP-1 | 6-7-fold at 48-72h | Similar to ALC0315 | High NF-κB activation |
| No ionizable lipid | No activation | No activation | Control confirmation |
The following diagram illustrates the key signaling pathways through which LNPs trigger inflammatory responses:
Strategic Approaches for Mitigation
Novel Ionizable Lipid Design
Advanced ionizable lipids with inherent anti-inflammatory properties represent a promising strategy:
- Hydroxychloroquine-functionalized lipids (HLs): Incorporate HCQ's anti-inflammatory properties directly into lipid structure, inhibiting endosomal TLRs (TLR3, TLR7, TLR9) and cGAS-STING signaling pathway [63]
- Antioxidant ionizable lipids (C-a16): Mitigate generation of intracellular reactive oxygen species (ROS), reducing inflammatory responses and prolonging protein expression duration [64]
- Performance benefits: C-a16 LNPs increase gene editing efficiency by 2.8× and protein expression by 3.6× compared to conventional LNPs [64]
mRNA Engineering and Modification
- Nucleoside modifications: Complete replacement of uridine with N1-methylpseudouridine (m1ψ) reduces RNA sensor activation and type 1 interferon responses [59]
- Sequence optimization: Codon optimization and UTR engineering to minimize secondary structures that activate immune sensors [11]
- Purification protocols: Rigorous removal of double-stranded RNA (dsRNA) impurities to reduce innate immune activation [59] [27]
LNP Formulation Optimization
- Component ratio adjustment: Systematic variation of ionizable lipid percentages (typically 35-50%) to balance delivery efficiency and immunogenicity [61]
- PEG-lipid selection: Optimization of PEG-lipid types and percentages (1-3%) to influence biodistribution and reduce immune recognition [11]
- Helper lipid screening: Evaluation of different phospholipids and cholesterol derivatives for reduced complement activation [11] [63]
Table 2: Quantitative Assessment of Mitigation Strategies
| Mitigation Strategy | Reduction in Cytokines | Impact on Protein Expression | Effect on Therapeutic Efficacy |
|---|---|---|---|
| m1ψ mRNA modification | IFN signaling reduced by >50% | Increased expression 40-46% | Enhanced vaccine immunogenicity |
| HCQ-functionalized lipids | Significant reduction in TNF-α, IFN-γ, IL-6 | Maintained expression levels | Improved safety in repeated dosing |
| Antioxidant C-a16 lipids | Reduced ROS-mediated inflammation | 3.6× increase in FGF21 expression | 2.8× higher gene editing efficiency |
| dsRNA removal | Antiviral gene signature reduced | Increased translational efficiency | Improved consistency of response |
Experimental Protocols
Protocol 1: In Vitro Immunogenicity Profiling
Purpose: Comprehensive assessment of innate immune activation by novel LNP formulations.
Materials:
- THP-1 monocyte cell line (NF-κB/IRF reporter systems)
- Primary human dendritic cells
- Cytokine measurement kits (IL-6, TNF-α, IFN-α, IFN-β, IL-1β)
- Puromycin incorporation assay reagents
- RNA sequencing reagents
Methodology:
Cell culture and stimulation:
- Maintain THP-1 cells in RPMI-1640 with 10% FBS
- Seed cells at 2×10⁵ cells/well in 24-well plates
- Stimulate with test LNPs at concentrations of 0.1-10 μg/mL for 4-48 hours
- Include controls: empty LNPs, mRNA-LNPs, TLR agonists (R848, MPLA)
NF-κB/IRF activation kinetics:
- Measure reporter activity at 4, 8, 24, 48, 72, and 120 hours post-stimulation
- Normalize to untreated controls and calculate fold-induction
- Generate dose-response curves at 48-hour timepoint
Cytokine profiling:
- Collect supernatants at 6, 24, and 48 hours
- Quantify IL-6, TNF-α, IFN-α, IFN-β, IL-1β via ELISA or multiplex assays
- Compare cytokine secretion patterns across formulations
Global translation assessment:
- Perform puromycin incorporation assay 20 hours post-transfection
- Treat cells with 10 μM puromycin for 15 minutes
- Fix cells and stain with anti-puromycin antibody
- Quantify fluorescence intensity via flow cytometry or imaging
Transcriptomic analysis:
- Extract total RNA at 1, 4, and 24 hours post-stimulation
- Perform RNA sequencing with 30 million reads per sample
- Conduct gene set variation analysis (GSVA) using MSigDB module C5
- Focus on antiviral response genes (OAS family, MX1, IFIT family)
Data Analysis:
- Calculate IC50 values for translational repression
- Determine peak activation times for NF-κB and IRF pathways
- Identify significantly enriched gene ontology terms in transcriptomic data
Protocol 2: In Vivo Tolerability and Efficacy Assessment
Purpose: Evaluate immunogenicity and therapeutic efficacy of novel LNP formulations in animal models.
Materials:
- C57BL/6 mice (6-8 weeks old)
- Test LNP formulations (0.1-1.0 mg/kg mRNA dose)
- Blood collection equipment (serum separation tubes)
- Tissue processing reagents for immunohistochemistry
- ELISA kits for murine cytokines
Methodology:
Study design:
- Randomize mice into groups (n=6-8 per formulation)
- Administer LNPs via intravenous or intramuscular injection
- Include vehicle control, conventional LNP, and test LNP formulations
- For repeated dosing studies, administer 3 doses at 2-week intervals
Systemic cytokine measurement:
- Collect blood via retro-orbital bleeding at 6, 24, and 48 hours post-injection
- Separate serum and store at -80°C
- Analyze IL-6, TNF-α, IFN-γ, IL-12, and KC/GRO levels via multiplex ELISA
Inflammatory cell infiltration:
- Euthanize animals 72 hours post-injection
- Collect injection site tissues (muscle for IM, liver for IV)
- Fix in 4% paraformaldehyde for 24 hours
- Process for H&E staining and immunohistochemistry
- Quantify neutrophil and macrophage infiltration per high-power field
Therapeutic efficacy assessment:
- Transfert with reporter mRNA (Luciferase or GFP)
- Image protein expression at 6, 24, 48, 72, and 96 hours
- Quantify expression intensity and duration
- For gene editing applications, assess editing efficiency via next-generation sequencing
Antigen-specific immune responses:
- Immunize with OVA or tumor antigen mRNA
- Measure antigen-specific T cells via ELISpot (IFN-γ) at 7 and 14 days
- Quantify antibody titers via ELISA at 14, 28, and 56 days
Data Analysis:
- Calculate area under curve for protein expression kinetics
- Compare peak cytokine levels and time to return to baseline
- Determine statistical significance via one-way ANOVA with post-hoc testing
The following workflow outlines the comprehensive screening approach for novel ionizable lipids:
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Immunogenicity Assessment
| Reagent/Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| Reporter Cell Lines | THP-1 NF-κB/IRF, HEK-Blue TLR4 | High-throughput screening of innate immune activation | Verify pathway specificity with knockout controls |
| Cytokine Assays | Luminex multiplex, ELISA kits | Quantification of inflammatory mediators | Measure multiple timepoints for kinetic analysis |
| Ionizable Lipids | SM-102, ALC-0315, C-a16, HCQ-lipids | LNP formulation components | Source from reputable suppliers with quality documentation |
| mRNA Constructs | Nucleoside-modified, unmodified, saRNA | Cargo for immunogenicity studies | Verify modification percentage via LC-MS |
| TLR Agonists/Antagonists | R848 (TLR7/8), MPLA (TLR4), HCQ | Controls and mechanism studies | Use EC50 concentrations for relevant comparisons |
The mitigation of LNP immunogenicity requires a multi-faceted approach addressing both the mRNA cargo and delivery vehicle. The development of novel ionizable lipids with inherent anti-inflammatory properties, combined with advanced mRNA engineering, presents a promising path toward safer LNP-based reprogramming mRNA therapeutics. The experimental protocols outlined herein provide a framework for comprehensive immunogenicity assessment during LNP development. As the field advances, the integration of these mitigation strategies will be crucial for expanding the application of LNP-mRNA platforms beyond vaccines to include sensitive therapeutic areas requiring repeated administration or precise dosing control.
Improving Biodegradability and Reducing Off-Target Effects
Lipid nanoparticles (LNPs) have emerged as the leading non-viral delivery vector for reprogramming mRNA, enabling revolutionary advances in gene editing and cellular reprogramming. However, two significant challenges impede their transition to safe and effective clinical therapies: insufficient biodegradability leading to potential long-term toxicity, and off-target effects that can compromise therapeutic precision. This Application Note provides detailed, actionable protocols to address these critical limitations, equipping researchers with methodologies to engineer next-generation LNPs with enhanced safety profiles for sensitive applications like reprogramming.
Strategic Approaches and Quantitative Comparison
Innovative strategies focusing on novel lipid chemistries and advanced targeting mechanisms are paving the way for safer LNP-based reprogramming platforms. The quantitative benefits of these approaches are summarized in Table 1 below.
Table 1: Quantitative Comparison of Strategies for Improving LNP Biodegradability and Reducing Off-Target Effects
| Strategy | Key Feature/Component | Reported Efficacy/Outcome | Primary Advantage | Relevant Protocol |
|---|---|---|---|---|
| Ester-Modified Ionizable Lipids [12] [65] | Cyclic structures with ester-functionalized tails | 100-fold higher potency than SM-102; Reduced liver expression [12] [65] | Enhanced biodegradability and endosomal escape | Protocol 3.1 |
| Mn2+-mRNA Core Enrichment [13] | Manganese ion condensed mRNA core | ~2-fold increase in mRNA loading capacity; 95.6% mRNA by weight in core [13] | Dose-sparing; reduces required lipid excipients | Protocol 3.2 |
| miR-122 Binding Site Insertion [66] | miRNA binding sites in mRNA UTRs | Significant reduction in off-target liver protein expression after intramuscular injection [66] | Passive de-targeting from liver | Protocol 3.3 |
| AI-Guided Lipid Design [67] | Machine learning & GANs for novel lipid design | Predicts lipid pKa with MAE <0.15; generates 92% novel ionizable lipids [67] | Accelerates discovery of biodegradable, targeted lipids | - |
| Optimized Buffer Formulations [68] | Mildly acidic histidine buffer | Enables room-temperature LNP stability for 6 months (vs. 2 weeks in PBS) [68] | Mitigates lipid oxidation and RNA-lipid adduct formation | - |
The following diagram illustrates the logical relationship and synergistic application of these core strategies within a development workflow.
Detailed Experimental Protocols
Protocol: Synthesis and Characterization of Ester-Modified Ionizable Lipids
This protocol describes the synthesis and screening of degradable cyclic amino alcohol ionizable lipids, such as the high-performing AMG1541, for potent mRNA delivery with reduced toxicity [12].
Materials:
- Reagents: Starting materials for lipid synthesis (e.g., epoxides, amines, acyl chlorides), anhydrous solvents (THF, DCM), mRNA (e.g., luciferase, antigen), commercial LNP components (DSPC, Cholesterol, DMG-PEG2000), formulation buffer (e.g., citrate, Tris).
- Equipment: Microfluidic mixer (e.g., NanoAssemblr), HPLC system, DLS/Zetasizer, TEM, cell culture suite, animal facility.
Procedure:
- Lipid Synthesis:
- Synthesize ionizable lipids containing cyclic amine headgroups and ester-functionalized hydrocarbon tails via nucleophilic ring-opening or amidation reactions [12].
- Critical Step: Purify the final product using flash chromatography to achieve >95% purity. Confirm structure via ( ^1H )-NMR and mass spectrometry.
LNP Formulation:
- Prepare an ethanolic lipid mixture containing the novel ionizable lipid, DSPC, cholesterol, and DMG-PEG2000 at a molar ratio of 50:10:38.5:1.5 [12].
- Prepare an aqueous phase containing the target mRNA in 50 mM citrate buffer, pH 4.0.
- Use a microfluidic mixer to combine the ethanolic and aqueous phases at a 1:3 volumetric flow rate ratio (ethanol:aqueous) to form LNPs [12] [68].
- Dialyze the formed LNPs against PBS or a suitable buffer (e.g., histidine) for 24 hours to remove residual ethanol.
In Vitro Characterization:
- Size and PDI: Determine hydrodynamic diameter and polydispersity index (PDI) via DLS. Acceptable PDI is ≤0.2 [68].
- Encapsulation Efficiency: Quantify using the Ribogreen assay. Centrifuge an LNP aliquot to pellet encapsulated mRNA. Compare fluorescence of the supernatant (free mRNA) to a lysed LNP sample (total mRNA). EE > 85% is target [68].
- Potency Assay: Transfert a relevant cell line (e.g., HEK293, dendritic cells) with LNPs encapsulating luciferase mRNA. Measure luminescence 24-48 hours post-transfection. Compare to benchmarks (e.g., SM-102 LNPs) [12].
In Vivo Biodistribution and Toxicity:
Protocol: Manganese Ion-Mediated High-Density mRNA Core Formation
This protocol details the creation of high-density mRNA cores using Mn2+ to significantly increase mRNA loading capacity, enabling dose-sparing and reduced lipid-related toxicity [13].
Materials:
- Reagents: mRNA, Manganese(II) chloride (MnCl2), Nuclease-free water, Lipids for coating (e.g., DOPE, DOTAP, ionizable lipids), Coomassie Blue protein assay reagent.
- Equipment: Thermomixer, Transmission Electron Microscope (TEM), Dynamic Light Scattering (DLS) instrument, Microfluidic mixer, Agarose gel electrophoresis system.
Procedure:
- Mn-mRNA Core Nanoparticle Formation:
- Dilute mRNA in nuclease-free water to a concentration of 0.1 mg/mL.
- Add an aqueous solution of MnCl2 to the mRNA solution to achieve a molar ratio of Mn2+ to mRNA bases of 5:1 [13].
- Incubate the mixture at 65°C for exactly 5 minutes in a thermomixer with mild agitation [13].
- Critical Step: Immediately cool the mixture on ice to halt the assembly process.
Lipid Coating (L@Mn-mRNA):
- Prepare an ethanolic lipid mixture suitable for your application (e.g., similar to the mixture in Protocol 3.1).
- Use a microfluidic mixer to combine the pre-formed Mn-mRNA nanoparticle suspension with the ethanolic lipid mix. This forms a lipid coat around the dense mRNA core [13].
- Dialyze the final L@Mn-mRNA particles into the desired storage buffer.
Characterization and Validation:
- mRNA Integrity: Verify mRNA integrity before and after Mn-mRNA core formation using agarose gel electrophoresis. No smearing or fragmentation should be visible [13].
- Nanoparticle Morphology: Use TEM to confirm the spherical morphology and core-shell structure of the final L@Mn-mRNA particles.
- Loading Capacity: Calculate the mRNA loading capacity as (mass of mRNA / total mass of LNP) * 100%. This formulation targets ~10% w/w, nearly double that of conventional LNPs [13].
- Functional Potency: Test the vaccine or therapeutic efficacy in a cellular uptake assay and/or an in vivo immunization model, comparing against conventional LNP-mRNA.
Protocol: Incorporating miRNA Binding Sites for Tissue-Specific Detargeting
This protocol outlines the use of microRNA-responsive binding sites in the mRNA construct to passively reduce off-target protein expression in specific tissues, such as the liver [66].
Materials:
- Reagents: Plasmid DNA template for in vitro transcription (IVT), miR-122 binding site oligonucleotides, IVT kit, HPLC purification kit, LNP formulation reagents.
- Equipment: Standard molecular biology suite (PCR machine, gel electrophoresis), IVT setup, Nanoparticle formulation equipment.
Procedure:
- mRNA Construct Design:
- Design a DNA template for your target reprogramming mRNA.
- Insert one to five tandem copies of the perfect complementary sequence to the 5' end of miR-122 (5'-UAACAGUCA-3') into the 3' untranslated region (3' UTR) of your mRNA construct [66].
- Note: The protocol indicates no significant difference in de-targeting efficacy between 1 or multiple sites, or between 5' and 3' UTR placement, offering flexibility [66].
mRNA Production and Purification:
- Perform in vitro transcription (IVT) from the linearized DNA template, incorporating base modifications like N1-methylpseudouridine to reduce immunogenicity.
- Purify the mRNA using HPLC or column-based methods to ensure high purity and integrity.
LNP Formulation and Testing:
- Formulate the modified mRNA into LNPs using standard procedures (see Protocol 3.1).
- In Vivo Validation:
- Systemically administer LNPs intravenously to mice to assess broad biodistribution.
- Alternatively, administer LNPs intramuscularly to model local delivery and assess systemic leakage.
- Quantify protein expression (e.g., via luciferase imaging or ELISA) in the target tissue (e.g., muscle) and the off-target liver tissue. Compare to a control group receiving LNPs with unmodified mRNA [66].
- Expect a significant reduction in liver expression for the miR-122-modified group with no significant impact on expression at the intramuscular injection site [66].
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for LNP Optimization and Characterization
| Reagent / Material | Function / Application | Key Considerations |
|---|---|---|
| Ionizable Lipids (e.g., AMG1541, SM-102) | Key component for mRNA encapsulation and endosomal escape; target for biodegradability engineering [12] [11]. | Prioritize lipids with hydrolysable bonds (e.g., esters). PKa should be ~6.2-6.8 for optimal endosomal escape [67]. |
| Manganese Chloride (MnCl₂) | Used to condense mRNA into high-density core nanoparticles (Mn-mRNA) for improved loading [13]. | Optimize Mn2+:base molar ratio (2:1 to 8:1). Strictly control heating (65°C for 5 min) to prevent mRNA degradation [13]. |
| DMG-PEG2000 | PEG-lipid; stabilizes LNP surface and prevents aggregation; influences pharmacokinetics and immunogenicity [11] [68]. | Molar percentage typically 1.5-2%. Can be a source of anti-PEG immunity; consider alternatives for repeated dosing. |
| Histidine Buffer | Optimized buffer for drug product matrix; mitigates lipid oxidation and significantly enhances room-temperature stability [68]. | Superior to phosphate buffers for long-term stability. Use at mildly acidic pH. |
| miR-122 Binding Site Oligos | DNA oligonucleotides encoding the binding site for liver-specific miR-122; cloned into mRNA UTRs to reduce off-target liver expression [66]. | A single site in the 3' UTR is sufficient for effective de-targeting. Ensure sequence is perfectly complementary. |
| RiboGreen Assay Kit | Fluorescent assay for highly sensitive quantification of RNA concentration; used to determine LNP encapsulation efficiency [13]. | Requires a "free RNA" measurement from an un-lysed LNP sample and a "total RNA" measurement from a lysed sample. |
Evaluating LNP Performance: Efficacy, Targeting Precision, and Clinical Potential
In lipid nanoparticle (LNP)-based reprogramming mRNA research, biophysical characterization provides the essential bridge between nanoparticle design and biological performance. The internal structure, size, surface characteristics, and physical stability of LNPs directly dictate their ability to successfully deliver reprogramming mRNA and modulate cellular protein production [69] [70]. For research focused on cellular reprogramming, where precise temporal control over transfected protein expression is critical, understanding these structure-function relationships becomes paramount [70] [71]. This Application Note details standardized protocols and analytical methodologies to systematically characterize LNP structural attributes and correlate them with functional outcomes in reprogramming applications, enabling researchers to design more effective mRNA delivery systems.
Key Biophysical Properties and Their Functional Implications
The structural and physical properties of mRNA-LNPs serve as critical quality attributes (CQAs) that significantly influence their in vitro and in vivo behavior. The following parameters are essential for evaluating LNP performance in reprogramming mRNA delivery.
Table 1: Essential Biophysical Properties of mRNA-LNPs and Their Functional Significance
| Biophysical Property | Analytical Techniques | Impact on LNP Function |
|---|---|---|
| Particle Size & Distribution | Dynamic Light Scattering (DLS), Nanoparticle Tracking Analysis (NTA), Multiangle DLS (MADLS) [72] | Influences cellular uptake, biodistribution, and transfection efficiency [72] [70]. |
| Surface Charge (Zeta Potential) | Electrophoretic Light Scattering (ELS) [72] | Predicts colloidal stability and interaction with cell membranes [72] [71]. |
| Internal Structure & Morphology | Cryo-Electron Microscopy (Cryo-EM), Small-Angle X-Ray Scattering (SAXS) [69] [70] | Determines mRNA encapsulation efficiency and release kinetics; linked to endosomal escape [69] [45]. |
| Particle Concentration & Encapsulation | MADLS, NTA, RiboGreen Assay [72] [73] | Informs dosing accuracy and yield; crucial for reproducible research [72] [73]. |
| Thermal Stability | Differential Scanning Calorimetry (DSC), DLS Thermal Ramp [72] | Indicates storage stability and shelf-life; essential for protocol planning [72]. |
The internal structure of LNPs, particularly the organization of mRNA and lipids, is a major determinant of functionality. Studies have identified distinct structural features, such as electron-dense cores with inverted hexagonal phases that facilitate efficient mRNA encapsulation and endosomal escape, which is critical for the cytosolic delivery of reprogramming factors [69] [70]. Furthermore, structural classifications like "eLNPs" (emulsion-like) and "mLNPs" (membrane-like) based on Cryo-EM analysis have been shown to correlate with different in vivo expression profiles and immune responses—a key consideration for sustained protein expression required in cellular reprogramming [69].
Diagram 1: LNP Structure-Function Workflow. This diagram outlines the logical relationship from LNP formulation through structural and functional analysis to the final reprogramming outcome.
Experimental Protocols for Comprehensive LNP Characterization
Protocol: Microfluidic Synthesis of mRNA-LNPs
Principle: This protocol describes a reproducible method for synthesizing LNPs using microfluidic mixing, which offers superior control over particle properties compared to traditional methods like pipette vortexing [73].
Materials:
- Lipids: Ionizable lipid (e.g., DLin-MC3-DMA, SM-102), phospholipid (e.g., DSPC), cholesterol, PEG-lipid (e.g., DMG-PEG2000).
- mRNA: Purified reprogramming mRNA (e.g., encoding transcription factors).
- Solvents: Ethanol (absolute), sodium acetate buffer (10 mM, pH 4.0).
- Equipment: Syringe pump, commercially available microfluidic chip (e.g., from Dolomite Microfluidics), syringes (1 mL), tubing, dialysis cassettes (e.g., MWCO 3.5 kDa).
Procedure:
- Lipid Solution: Dissolve the lipid mixture (ionizable lipid, DSPC, cholesterol, PEG-lipid at a molar ratio of 50:10:38.5:1.5) in ethanol to a final concentration of 10-12.5 mg/mL total lipids. Gently warm if necessary to dissolve completely [73] [74].
- mRNA Solution: Dilute the reprogramming mRNA in sodium acetate buffer (pH 4.0) to a concentration of 50-200 μg/mL.
- Microfluidic Mixing:
- Load the lipid-ethanol solution and mRNA-aqueous solution into separate syringes.
- Connect the syringes to the microfluidic chip via tubing and place them on the syringe pump.
- Set the total flow rate (TFR) to 10-12 mL/min with a aqueous-to-ethanol flow rate ratio (FRR) of 3:1.
- Initiate pumping to mix the streams turbulently within the chip, resulting in instantaneous LNP formation [73].
- Dialysis and Buffer Exchange:
- Collect the LNP suspension and dialyze against a large volume of PBS (pH 7.4) for at least 4 hours at 4°C to remove ethanol and adjust the pH. Alternatively, use Tangential Flow Filtration (TFF) for more controlled buffer exchange and concentration [74].
- Sterile-filter the final formulation (0.22 μm pore size) and store at 4°C.
Notes: The TFR and FRR can be adjusted to fine-tune LNP size. The protocol has been validated for user-to-user reproducibility, even with novice operators [73].
Protocol: Multi-Technique Biophysical Characterization
Principle: A combination of complementary techniques is required to obtain a holistic understanding of LNP physical attributes, as no single method provides all necessary information [72].
Materials:
- Purified LNP suspension.
- DLS/SLS Instrument: e.g., Zetasizer Ultra (Malvern Panalytical).
- NTA Instrument: e.g., NanoSight NS300 (Malvern Panalytical).
- ELS Instrument: Typically integrated in the DLS system.
- Fluorescence Spectrophotometer and Quant-iT RiboGreen RNA Reagent kit.
Procedure:
- Particle Size, PDI, and Concentration (DLS/MADLS):
- Dilute the LNP suspension in PBS to achieve an appropriate scattering intensity.
- Load into a disposable microcuvette and measure using DLS to determine the hydrodynamic diameter (Z-average) and polydispersity index (PDI).
- For higher-resolution size distribution and particle concentration, use MADLS, which combines measurements from multiple angles [72].
Particle Concentration and Size (NTA):
- Dilute LNPs in filtered PBS to visualize approximately 20-100 particles per frame.
- Inject the sample into the instrument and record five 60-second videos.
- Analyze the videos to determine the particle concentration (particles/mL) and the mode of the size distribution based on direct particle tracking [72].
Zeta Potential (ELS):
- Dilute LNPs in 1 mM KCl or 10 mM NaCl to ensure a low conductivity.
- Load the sample into a folded capillary zeta cell.
- Measure the electrophoretic mobility and use the Smoluchowski model to calculate the zeta potential, which indicates surface charge [72].
mRNA Encapsulation Efficiency (RiboGreen Assay):
- Prepare two sets of LNPs diluted in TE buffer (1:1000).
- To one set, add 2% Triton X-100 to disrupt the LNPs (Total RNA sample).
- Leave the other set intact (Encapsulated RNA sample).
- Add RiboGreen dye to both sets and measure fluorescence (excitation: ~480 nm, emission: ~520 nm).
- Calculate encapsulation efficiency (%) as:
[1 - (Encapsulated RNA Fluorescence / Total RNA Fluorescence)] × 100[73].
Table 2: Troubleshooting Common LNP Characterization Issues
| Problem | Potential Cause | Solution |
|---|---|---|
| High PDI (>0.2) | Inconsistent mixing during synthesis, aggregation | Optimize flow rates in microfluidics; ensure fresh buffer for dialysis; filter before measurement [73]. |
| Low Encapsulation Efficiency | Incorrect N/P ratio, rapid mixing | Verify lipid and mRNA input calculations; adjust TFR/FRR to optimize self-assembly [73] [74]. |
| Large Particle Size (>150 nm) | High lipid concentration, aggregation | Dilute lipid stock prior to mixing; check for impurities in buffers or mRNA [73]. |
| Negative Zeta Potential | PEG-lipid outer layer, mRNA on surface | This is typical for stable, stealth LNPs. A highly positive charge may indicate toxicity issues [72] [71]. |
The Scientist's Toolkit: Essential Reagents and Materials
Successful characterization requires specific reagents and instruments. The following table lists key solutions for standard LNP biophysical analysis.
Table 3: Research Reagent Solutions for LNP Characterization
| Research Reagent / Material | Function / Role in Characterization |
|---|---|
| Ionizable Cationic Lipids (e.g., DLin-MC3-DMA, SM-102) | Core component for mRNA encapsulation and endosomal escape; its structure dictates LNP efficacy [11] [74]. |
| Phospholipids (e.g., DSPC, DOPE) | Helper lipids that provide structural integrity to the LNP bilayer and can influence fusogenicity [11] [71]. |
| Cholesterol | Enhances the stability and fluidity of the lipid bilayer and promotes membrane fusion for endosomal escape [11] [71]. |
| PEGylated Lipids (e.g., DMG-PEG2000) | Shields LNP surface, reduces aggregation, controls particle size, and impacts pharmacokinetics [11] [74]. |
| Quant-iT RiboGreen Assay | Fluorescent dye used to accurately quantify both encapsulated and total mRNA, critical for determining encapsulation efficiency [73]. |
| Dynamic Light Scattering (DLS) Instrument | Workhorse instrument for measuring hydrodynamic diameter, polydispersity (PDI), and overall size distribution of LNPs [72]. |
| Nanoparticle Tracking Analysis (NTA) Instrument | Provides direct visualization and measurement of particle concentration and size distribution based on Brownian motion [72]. |
Robust biophysical characterization is not merely a quality control step but a fundamental practice for elucidating the structure-function relationships of mRNA-LNPs. By implementing the standardized protocols and multi-technique approaches outlined in this Application Note, researchers can systematically optimize LNP formulations for cellular reprogramming applications. This data-driven strategy enables the rational design of next-generation LNPs with enhanced efficacy and safety profiles, accelerating the development of advanced mRNA-based therapies.
The advent of lipid nanoparticle (LNP)-delivered reprogramming messenger RNA (mRNA) represents a paradigm shift in therapeutic development, enabling transient, in situ production of proteins for applications ranging from regenerative medicine to cancer immunotherapy [75] [62]. The clinical success of LNP-based mRNA vaccines during the COVID-19 pandemic demonstrated the viability of this platform, with Pfizer/BioNTech's BNT162b2 and Moderna's mRNA-1273 achieving efficacy rates of 95% and 94.1%, respectively [34]. These milestones underscore the critical importance of robust in vivo efficacy models in translating promising laboratory findings into clinical therapeutics.
A comprehensive understanding of LNP-mRNA pharmacology is essential for designing predictive efficacy models. This includes characterizing the hierarchical biological trajectory of mRNA-LNP formulations, from initial systemic exposure and tissue-specific biodistribution to intracellular delivery and ultimate protein expression dynamics [76]. Despite their success, fundamental knowledge gaps remain in comprehensively defining this trajectory, posing significant constraints on the rational design of next-generation mRNA-LNP therapeutics [76]. This document provides a detailed framework of in vivo efficacy models, experimental protocols, and key considerations for evaluating LNP-delivered reprogramming mRNA, with a focus on bridging preclinical findings to clinical outcomes.
LNP-mRNA Pharmacology and Kinetic Considerations
The in vivo fate of LNP-mRNA formulations is governed by a complex interplay of biological processes. Systemically administered LNPs encounter biological fluids where proteins spontaneously adsorb to form a "protein corona" that redefines their physicochemical properties and influences delivery outcomes [77]. This corona composition affects cellular uptake, biodistribution, and ultimately, transfection efficiency. Surprisingly, increased cellular uptake mediated by certain corona proteins does not necessarily correlate with enhanced mRNA expression, potentially due to protein corona-induced alterations in intracellular trafficking, such as lysosomal sequestration [77].
Endosomal escape represents a critical bottleneck in LNP-mRNA delivery efficiency. Current estimates suggest that less than 2-3% of nucleic acids successfully escape the endosome and are released into the cytosol [22]. This limitation highlights the importance of LNP composition, particularly ionizable lipids that undergo charge transitions in acidic endosomal environments, interacting with anionic phospholipids to facilitate membrane fusion and cargo release [22].
Table 1: Key Pharmacokinetic Parameters of LNP-mRNA Formulations
| Parameter | Description | Impact on Efficacy | Measurement Methods |
|---|---|---|---|
| Biodistribution | Tissue-specific accumulation of LNPs and their components | Determines site of protein expression and potential off-target effects | Whole-body imaging, LC-MS/MS of tissue homogenates [76] |
| Cellular Uptake | Internalization of LNPs into target cells | Necessary but not sufficient for protein expression | Flow cytometry, fluorescence microscopy [77] |
| Endosomal Escape | Release of mRNA from endosomes into cytosol | Critical rate-limiting step (~2% efficiency) [22] | Fluorescent dye-based assays, electron microscopy |
| Protein Expression Kinetics | Onset, magnitude, and duration of encoded protein production | Determines therapeutic dosing regimen | Bioluminescence imaging, ELISA, Western blot [78] |
| Clearance | Elimination of LNP components and mRNA degradation products | Affects duration of exposure and potential toxicity | Mass spectrometry, radioactive tracing [76] |
The translational efficiency of mRNA is influenced by multiple design elements, including 5' capping, 5' and 3' untranslated regions (UTRs), nucleotide modification (e.g., N1-methylpseudouridine, m1Ψ), and poly(A) tail length [78] [62]. These elements collectively impact mRNA stability, translational capacity, and immunogenicity, all of which must be optimized in the context of the specific therapeutic application.
Animal Models for Efficacy Assessment
Small Animal Models
Rodent models, particularly mice and rats, serve as the foundation for initial in vivo efficacy testing due to their manageable size, well-characterized genetics, and availability of disease models.
Immunocompetent Models: Syngeneic models using mice with intact immune systems are essential for evaluating immunomodulatory therapies, including cancer vaccines and infectious disease applications. For example, in cancer immunotherapy development, syngeneic models allow assessment of both direct antitumor effects and activation of host immune responses [62].
Disease-Specific Models: Transgenic, knockout, and humanized mouse models recapitulate specific human diseases. For metabolic disorders like methylmalonic acidemia, mRNA-3704 (encoding methylmalonyl-CoA mutase) has been evaluated in phase I/II clinical trials following promising preclinical results in disease models [79].
Reprogramming Models: Direct in vivo reprogramming of resident cells represents a powerful application of LNP-mRNA technology. Cardiac reprogramming studies have demonstrated that combinations of transcription factors (e.g., Gata4, Mef2c, Tbx5) or specific microRNAs (miR-1, miR-133, miR-208, miR-499) can convert fibroblasts to cardiomyocyte-like cells in situ, offering potential for regenerative therapy [75].
Figure 1: In Vivo Efficacy Evaluation Workflow. This diagram outlines the key stages in preclinical assessment of LNP-mRNA therapeutics, from formulation to data analysis.
Large Animal Models
Large animal models, including non-human primates, pigs, and dogs, provide critical translational bridges to human clinical trials. Their physiological and anatomical similarities to humans, particularly in cardiovascular and immune systems, offer more predictive assessment of dosing, biodistribution, and potential toxicities. The LNP-mRNA vaccines against SARS-CoV-2 underwent extensive testing in non-human primates prior to human trials, establishing proof-of-concept for protection against viral challenge and informing dose selection [34].
Quantitative Efficacy Endpoints
Biodistribution and Protein Expression
Biodistribution studies quantify the tissue-specific accumulation of LNPs and their mRNA cargo, while expression analysis measures the resulting functional protein output.
Protocol 4.1: Quantitative Biodistribution and Expression Analysis
Materials:
- LNP-mRNA formulation
- Appropriate animal model
- In vivo imaging system (for luminescent/fluorescent reporters)
- Tissue homogenization equipment
- RNA extraction kit
- qRT-PCR system
- ELISA materials for target protein detection
Procedure:
- Administer LNP-mRNA formulation via intended route (e.g., intravenous, intramuscular) at predetermined dose.
- At designated time points (e.g., 6h, 24h, 48h, 72h, 1 week), euthanize animals and collect tissues of interest (liver, spleen, lung, heart, muscle, injection site).
- For biodistribution: Homogenize tissues and extract total RNA. Perform qRT-PCR using probes specific to the administered mRNA.
- For protein expression: Process tissues for protein extraction or perform in situ hybridization/immunohistochemistry. Alternatively, use ELISA for quantitative measurement.
- Analyze data relative to control groups (e.g., saline, empty LNPs).
Data Interpretation: Normalize mRNA levels to housekeeping genes and protein expression to total protein content. Calculate tissue-to-plasma ratios and compare expression kinetics across time points.
Table 2: Comparison of LNP Formulations for Intramuscular mRNA Delivery [78]
| Ionizable Lipid | LNP Size (nm) | Encapsulation Efficiency (%) | Relative Luciferase Expression | Stability at 4°C |
|---|---|---|---|---|
| SM-102 (Moderna) | 75.5 ± 0.4 | >95 | 160% | High |
| ALC-0315 (Pfizer-BioNTech) | 90.2 ± 7.8 | >95 | 100% | Moderate |
| cKK-E12 (Research) | 88.2 ± 1.5 | >95 | 105% | Moderate |
Functional Efficacy Endpoints
Functional assessments vary by therapeutic application but provide the most clinically relevant measures of activity.
Protocol 4.2: Tumor Growth Inhibition in Oncology Models
Materials:
- Syngeneic or xenograft tumor model
- Calipers or in vivo imaging system
- LNP-mRNA encoding immunomodulators or tumor antigens
- Flow cytometry equipment for immune cell profiling
Procedure:
- Implant tumor cells subcutaneously or orthotopically in appropriate mouse model.
- Randomize animals into treatment groups when tumors reach predetermined volume (typically 50-100 mm³).
- Administer LNP-mRNA formulations according to treatment schedule.
- Measure tumor dimensions 2-3 times weekly using calipers (volume = ½ × length × width²).
- Monitor body weight as general health indicator.
- At endpoint, harvest tumors and lymphoid organs for immune cell profiling by flow cytometry.
Data Interpretation: Calculate tumor growth inhibition (%) relative to control group. Perform statistical analysis on tumor volumes and survival curves. Evaluate changes in immune cell populations in tumor microenvironment.
Protocol 4.3: Functional Assessment in Cardiac Reprogramming Models
Materials:
- Myocardial infarction model
- Echocardiography system
- LNP-mRNA encoding reprogramming factors (e.g., GMT cocktail: Gata4, Mef2c, Tbx5)
- Histology materials
Procedure:
- Induce myocardial infarction via coronary artery ligation in mouse model.
- Administer LNP-mRNA formulations directly to infarct border zone or systemically.
- Perform serial echocardiography at baseline and weekly post-treatment to assess cardiac function (ejection fraction, fractional shortening).
- Monitor arrhythmia incidence via electrocardiography.
- At endpoint, process heart tissue for histology (fibrosis assessment, immunofluorescence for cardiomyocyte markers).
Data Interpretation: Compare functional parameters between treatment and control groups. Quantify extent of fibrosis and presence of reprogrammed cardiomyocytes in infarct region.
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for LNP-mRNA Research
| Reagent Category | Specific Examples | Function | Considerations |
|---|---|---|---|
| Ionizable Lipids | SM-102, ALC-0315, DLin-MC3-DMA [22] [78] | mRNA complexation, endosomal escape | SM-102 shows superior intramuscular delivery vs. ALC-0315 [78] |
| Helper Lipids | DSPC, DOPE, Cholesterol [22] | Structural support, membrane fusion | DOPE enhances endosomal escape; DSPC provides stability [22] |
| PEGylated Lipids | DMG-PEG2000, ALC-0159 [22] [78] | Stability, circulation time, "stealth" properties | PEG density affects protein corona formation and pharmacokinetics [77] |
| mRNA Modifications | N1-methylpseudouridine (m1Ψ) [78] [62] | Reduced immunogenicity, enhanced stability | Moderna and Pfizer-BioNTech use different m1Ψ content due to codon optimization [78] |
| UTR Optimizations | Pfizer-BioNTech 5' UTR, Moderna 3' UTR [78] | Enhanced translation efficiency | Specific UTR combinations significantly impact protein expression [78] |
Pathway to Clinical Trials
Translating preclinical findings to clinical success requires careful consideration of several key factors.
Dose Translation and Regimen Optimization
Species-specific differences in metabolism, immune function, and target biology complicate direct dose extrapolation. Allometric scaling based on body surface area provides an initial estimate, but LNP-specific factors such as opsonization, protein corona formation, and tissue tropism must be considered [77]. The dosing regimen (single vs. multiple administrations) should reflect the therapeutic goals—single doses may suffice for vaccines, while chronic conditions may require repeated administration.
Figure 2: Clinical Translation Pathway. This diagram outlines the key stages in translating LNP-mRNA therapeutics from preclinical models to clinical trials.
Clinical Endpoint Selection
Clinical trial endpoints should logically extend from the preclinical efficacy models. For LNP-mRNA vaccines, immunogenicity (neutralizing antibody titers, T-cell responses) serves as a primary initial endpoint, with prevention of infection or disease as the definitive clinical outcome [34]. For protein replacement therapies, biochemical correction (e.g., enzyme activity) and functional improvement represent appropriate endpoints. In regenerative applications, imaging-based assessments of tissue repair and functional measures are most relevant.
Safety and Immunogenicity
The inherent immunostimulatory properties of mRNA present both opportunities and challenges. In vaccine applications, immune activation is desirable, while in protein replacement or regenerative therapies, it may be detrimental [62]. Preclinical models should include comprehensive assessment of innate immune activation (elevated cytokines, inflammatory cell infiltration) and potential autoimmune reactions. Recent advances in nucleotide modification (e.g., m1Ψ) have substantially reduced immunogenicity while maintaining translational efficiency [78] [62].
Robust in vivo efficacy models are indispensable for advancing LNP-delivered reprogramming mRNA therapeutics from preclinical research to clinical application. The framework presented herein emphasizes comprehensive pharmacological characterization, disease-relevant functional endpoints, and thoughtful translation of findings across species. As the field evolves, continued refinement of these models—particularly through incorporation of humanized systems and advanced imaging technologies—will enhance their predictive value and accelerate the development of this promising therapeutic modality.
Lipid nanoparticles (LNPs) have emerged as the cornerstone delivery platform for messenger RNA (mRNA)-based therapeutics, as demonstrated by their pivotal role in the successful deployment of COVID-19 vaccines [19] [21]. Their application, however, extends far beyond prophylactic vaccines into the promising realm of regenerative medicine and cellular reprogramming. For researchers aiming to deliver reprogramming mRNAs—which instruct cells to adopt new identities or functions—the precise formulation of LNPs is paramount. The ionizable lipid, as the core functional component, alongside structural lipids like phospholipids and cholesterol, and stabilizing PEG-lipids, collectively determine the fate of the encapsulated mRNA [80] [81]. The delicate balance of these components dictates critical outcomes: the efficiency of mRNA delivery to the target cell, the specificity of this delivery to avoid off-target effects, and the overall safety profile of the nanocarrier [53] [21]. This application note provides a comparative analysis of modern LNP formulations, summarizing quantitative data and detailing standardized protocols to support their development for advanced reprogramming mRNA research.
Comparative Performance of LNP Formulations
The efficacy, specificity, and safety of LNPs are highly dependent on their individual lipid components. The data from recent studies, summarized in the table below, highlight how variations in lipid structure and composition lead to divergent experimental outcomes.
Table 1: Impact of Lipid Composition on LNP Performance
| Lipid Component & Variation | Key Experimental Finding | Implication for Reprogramming mRNA Research |
|---|---|---|
| Ionizable Lipid: Tail Length [53] | Lipid 7 (optimized tail) showed a 3-fold higher mRNA expression at injection site vs. benchmarks, while minimizing liver accumulation. | Enhances local protein expression crucial for in situ reprogramming while reducing hepatotoxicity. |
| Ionizable Lipid: Stereochemistry [21] | Stereopure C12-200-S LNPs delivered up to 6.1-fold more mRNA in vivo than their racemic counterparts. | Highlights the critical, often overlooked, role of chiral purity in designing ionizable lipids for high-fidelity delivery. |
| Cholesterol: Hydroxylation [21] | Substituting 25-50% of cholesterol with 7α-hydroxycholesterol improved mRNA delivery efficiency by 1.8 to 2.0-fold in primary human T cells. | Modifying sterol composition can enhance endosomal escape, a major barrier for efficient reprogramming. |
| PEG-Lipid: Molar Ratio [81] | Increasing PEG-lipid from 0% to 0.5% mol reduced LNP size from ~200 nm to 100 nm, improving consistency and stability. | Allows precise control over nanoparticle size, a key factor in biodistribution and cellular uptake. |
The strategic modification of the ionizable lipid's hydrophobic tail is a primary lever for controlling LNP behavior. Research by Jallow et al. emphasizes that shorter lipid tails may enhance potency for larger mRNAs, while unsaturated tails can facilitate membrane fusion and promote endosomal escape [80]. A seminal 2025 study directly demonstrated this by engineering a library of ionizable lipids with varying tail lengths, leading to the identification of "Lipid 7." This optimized LNP achieved a threefold increase in local mRNA expression and significantly reduced liver accumulation compared to conventional SM-102-based LNPs. This shift in biodistribution not only enhanced efficacy at the target site but also mitigated the risk of hepatotoxicity, a common concern with first-generation LNPs [53].
Beyond the ionizable lipid, other components can be fine-tuned. The substitution of cholesterol with modified versions like 7α-hydroxycholesterol has been shown to alter endosomal trafficking, reducing recycling and enhancing mRNA delivery efficiency by up to 2.0-fold in primary cells [21]. Furthermore, the molar ratio of PEG-lipid, though typically small, is critical for controlling LNP size and preventing aggregation, which directly impacts stability and in vivo circulation time [80] [81].
Table 2: Analytical Methods for Critical Quality Attributes (CQAs) of LNPs
| Critical Quality Attribute (CQA) | Recommended Analytical Technique | Key Methodological Insight |
|---|---|---|
| Particle Size & Polydispersity | Dynamic Light Scattering (DLS) | Target size range for intramuscular injection: 50-200 nm; PDI < 0.2 indicates a monodisperse population [53] [81]. |
| Lipid & Nucleic Acid Quantification | Reverse-Phase HPLC with ELSD/UV | Use a monolithic C4 column and TEAA buffer in an isopropanol gradient to separate and quantify all lipid components and mRNA in a single run [82]. |
| Encapsulation Efficiency | RiboGreen Fluorescence Assay | Measure fluorescence of mRNA in LNP samples before and after disruption with Triton X-100. EE% = [(Total mRNA - Free mRNA)/Total mRNA] x 100 [53]. |
| Structural Morphology | Cryo-Electron Microscopy (cryo-EM) | Visualizes lamellarity, core architecture, and lipid layer integrity, correlating structure with stability and release kinetics [80]. |
Experimental Protocols for LNP Development and Analysis
Protocol: Microfluidic Formulation of LNPs for Reprogramming mRNA
This protocol is adapted from industry-standard methods used in LNP production [53] [81].
Principle: LNPs are formed via rapid nanoprecipitation achieved by mixing a lipid solution in ethanol with an aqueous mRNA buffer in a microfluidic device. This ensures reproducible, monodisperse nanoparticles with high encapsulation efficiency.
Materials:
- Lipids: Ionizable lipid (e.g., SM-102, proprietary Lipid 7), DSPC, Cholesterol, DMG-PEG2000.
- mRNA: Purified, modified mRNA (e.g., pseudouridine) resuspended in nuclease-free water.
- Buffers: Ethanol (100%), 25 mM sodium acetate buffer (pH 4.0), Tris-HCl buffer (pH 7.4), PBS with 10% sucrose (storage buffer).
- Equipment: Microfluidic mixer (e.g., NanoAssemblr Ignite), syringes, tubing, tangential flow filtration (TFF) system or dialysis cassettes.
Procedure:
- Prepare Lipid Stock Solution: Dissolve ionizable lipid, DSPC, cholesterol, and DMG-PEG2000 in ethanol at a molar ratio of, for example, 50:10:38.5:1.5. Adjust the ratio of the ionizable lipid based on screening results (e.g., 45 mol% as in optimized Lipid 7 formulations) [53].
- Prepare Aqueous mRNA Solution: Dilute the reprogramming mRNA in 25 mM sodium acetate buffer (pH 4.0) to a final concentration suitable for the desired N/P ratio (typically 6-12).
- Microfluidic Mixing:
- Load the lipid-ethanol solution and the mRNA aqueous solution into separate syringes.
- Set the flow rate ratio to 3:1 (aqueous:ethanol) and the total flow rate to achieve rapid mixing (e.g., 12 mL/min total).
- Mix the streams in the microfluidic cartridge and collect the resulting LNP suspension in a receiving vessel.
- Buffer Exchange and Purification:
- Use TFF or dialysis against Tris-HCl buffer (pH 7.4) to remove ethanol and exchange the external buffer.
- Finally, exchange the buffer into PBS with 10% sucrose for stable storage at -80°C.
- Characterization: Determine particle size, PDI, and zeta potential by DLS. Measure encapsulation efficiency using the RiboGreen assay [53].
Protocol: Quantitative Lipid Analysis via RP-HPLC-ELSD
This protocol is critical for ensuring the consistency of LNP composition during development and manufacturing [82].
Principle: Lipids are separated based on hydrophobicity using a reverse-phase monolithic column and detected by an evaporative light scattering detector (ELSD), which is ideal for analytes lacking chromophores.
Materials:
- HPLC System: PATfix or equivalent system with quaternary pump, autosampler, and column oven.
- Detectors: ELSD (e.g., SEDEX LT-ELSD), multi-wavelength UV-Vis detector.
- Column: CIMac C4 HLD monolithic analytical column (0.1 mL volume).
- Mobile Phase: A) 10 mM Triethylammonium Acetate (TEAA) in water, B) Isopropanol (IPA).
- Standards: Pure standards of all lipid components (ionizable lipid, DSPC, Cholesterol, DMG-PEG2000).
Procedure:
- Sample Preparation: Dilute the LNP formulation with a 3:1 mixture of 10 mM TEAA and IPA. No disassembly of LNPs is required prior to injection.
- Chromatographic Conditions:
- Column Temperature: 30°C
- ELSD Evaporation Temperature: 53°C
- Injection Volume: 500 µL
- Gradient: Begin with 40% B, ramp to 100% B over 10 minutes, hold for 2 minutes.
- Flow Rate: 1.0 mL/min.
- Analysis: Identify lipids based on retention time compared to pure standards. The typical elution order is PEG-lipid, phospholipid, cholesterol, and ionizable/cationic lipid. Quantify the concentration of each lipid component using calibrated standard curves [82].
Visualization of LNP Workflow and Mechanism
The following diagrams, generated using Graphviz, illustrate the key processes in LNP development and their mechanism of action.
Diagram 1: LNP Screening Workflow
Diagram 2: LNP mRNA Delivery Mechanism
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for LNP-based Reprogramming mRNA Research
| Reagent / Material | Function / Role | Example & Notes |
|---|---|---|
| Ionizable Lipids | Core component; binds mRNA, enables endosomal escape. | SM-102: Benchmark lipid. Proprietary Lipids (e.g., Lipid 7): Engineered for reduced liver accumulation [53]. |
| Helper Phospholipids | Supports LNP bilayer structure and stability. | DSPC: Common choice for structural integrity. DOPE: Can promote membrane fusion [21]. |
| Sterols | Modulates membrane fluidity and stability. | Cholesterol: Standard choice. Hchol (Modified Cholesterol): Can enhance endosomal escape via pH-sensitive protonation [21]. |
| PEGylated Lipids | Controls LNP size, reduces aggregation, improves stability. | DMG-PEG2000: Commonly used; note potential for immunogenicity with repeated dosing [80] [21]. |
| Microfluidic System | Enables reproducible, scalable LNP formulation. | NanoAssemblr Ignite: Industry standard for research-scale production [82] [81]. |
| Analytical Chromatography | Quantifies lipid composition and purity. | RP-HPLC with ELSD: Uses monolithic C4 columns for efficient separation of all LNP components [82]. |
Lipid nanoparticles (LNPs) represent the leading non-viral delivery platform for reprogramming mRNA, a revolutionary approach with transformative potential in regenerative medicine and cell fate manipulation. The clinical success of mRNA vaccines during the COVID-19 pandemic has established two ionizable lipids, SM-102 (Moderna's mRNA-1273) and ALC-0315 (Pfizer-BioNTech's BNT162b2), as benchmark standards. For research aimed at translating in vitro reprogramming strategies into viable therapies, a critical understanding of these lipids' distinct performance characteristics is paramount. These lipids are integral to overcoming the fundamental delivery challenges associated with mRNA, including enzymatic degradation, cellular uptake, and efficient endosomal escape. This document provides a detailed, evidence-based comparison of SM-102 and ALC-0315 and outlines standardized experimental protocols for their evaluation in the context of reprogramming mRNA delivery, providing a foundational framework for research and development.
Head-to-Head Benchmarking of SM-102 and ALC-0315
Direct comparative studies offer the most valuable insights for lipid selection. A systematic in vivo investigation delved into the performance of LNPs formulated with these two benchmark lipids.
2.1 Key Comparative Data The table below summarizes quantitative findings from a controlled study comparing SM-102 and ALC-0315 LNPs encapsulating firefly luciferase (Fluc) mRNA, administered via intramuscular injection in mice [83].
Table 1: Direct Comparison of SM-102 and ALC-0315 LNP Performance
| Parameter | SM-102 | ALC-0315 | Experimental Context |
|---|---|---|---|
| Particle Size (nm) | 75.5 ± 0.4 | 90.2 ± 7.8 | As measured by dynamic light scattering (DLS) [83]. |
| Polydispersity Index (PDI) | Narrower distribution | Wider distribution | Indicates more homogeneous particle size for SM-102 LNPs [83]. |
| Encapsulation Efficiency | >95% | >95% | Both lipids demonstrate excellent mRNA encapsulation [83]. |
| In Vivo Protein Expression | ~60% higher | Baseline | Measured by bioluminescent imaging 24 hours post-intramuscular injection [83]. |
| Stability at 4°C | Superior | Inferior | SM-102 LNPs demonstrated better long-term stability [83]. |
| pKa | ~6.75 [84] | ~6.09 [84] | Impacts endosomal escape efficiency and ionizable behavior. |
| Primary Metabolite | Ester hydrolysis products [84] | Stereoisomers (S,S), (R,R), meso [85] | Influences safety and clearance profiles; (S,S)-ALC-0315 shows reduced toxicity [85]. |
| Ideal Application | mRNA vaccines & protein replacement [84] | Vaccines & immunotherapy [84] | SM-102 offers a balanced profile; ALC-0315 favors strong immunogenicity. |
2.2 Interpretation of Benchmarking Data The data indicates that while both lipids form stable, highly efficient LNPs, SM-102 may offer advantages in specific contexts. Its smaller particle size and narrower distribution can influence biodistribution and cellular uptake. The significantly higher in vivo protein expression is critical for reprogramming applications, where the efficiency of transcription factor translation directly impacts the yield of successfully reprogrammed cells. Furthermore, superior stability simplifies logistics for long-term research use. The recent discovery that the individual stereoisomers of ALC-0315 exhibit differing toxicity profiles opens new avenues for optimizing the safety of LNP formulations, with the (S,S)-isomer showing particular promise [85].
Detailed Experimental Protocols
This section provides a reproducible methodology for formulating and benchmarking LNP performance, based on published procedures [83] [86].
3.1 Protocol: Microfluidic Formulation of LNPs This protocol describes the preparation of LNPs using a microfluidic mixer, such as the NanoAssemblr Ignite, for highly reproducible particle synthesis.
- Objective: To prepare sterile, monodisperse LNPs encapsulating reprogramming mRNA using SM-102 and ALC-0315.
- Materials:
- Ionizable Lipids: SM-102 (Taskcm, #TC-SM102) or ALC-0315 (Taskcm, #TC-ALC0315).
- Helper Lipids: Cholesterol (Avanti, #700100), DSPC (Avanti, #850365P), DMG-PEG2000 (Avanti, #880151P).
- Aqueous Phase: 20 mM Tris/4.3 mM Acetate/10% sucrose (TAS) buffer, pH 7.4 [86].
- Organic Phase: Ethanol, USP grade.
- mRNA: Purified, modified (e.g., N1-methylpseudouridine) mRNA encoding reprogramming factors.
- Procedure:
- Lipid Stock Preparation: Dissolve the lipid components in ethanol to form the organic phase at a molar ratio of 50:10:38.5:1.5 (Ionizable Lipid:DSPC:Cholesterol:DMG-PEG2000) [86]. Total lipid concentration should be 5-10 mM.
- mRNA Preparation: Dilute the reprogramming mRNA in the TAS buffer (aqueous phase) to a concentration of 0.1 mg/mL.
- Mixing: Load the organic and aqueous phases into separate syringes. Use a microfluidic mixer with a fixed total flow rate (e.g., 12 mL/min) and a flow rate ratio (Aqueous:Organic) of 3:1 to rapidly mix the streams.
- Dialyzing/Buffer Exchange: Immediately transfer the resulting LNP mixture into a dialysis cassette (e.g., 20kD MWCO) and dialyze against a large volume of TAS buffer for 4 hours at 4°C to remove ethanol. Alternatively, use tangential flow filtration (TFF).
- Sterile Filtration: Filter the final LNP formulation through a 0.22 µm sterile filter.
- Characterization: Measure particle size, PDI, and zeta potential using dynamic light scattering (DLS). Determine encapsulation efficiency using a Ribogreen assay [83] [86].
The following workflow diagram illustrates the LNP formulation and characterization process.
3.2 Protocol: Evaluating Transfection Efficiency and Cytotoxicity This protocol assesses the functional delivery of mRNA and the safety of the LNP formulations in vitro.
- Objective: To quantify the protein expression and cytotoxicity of SM-102 and ALC-0315 LNPs in target cells.
- Materials:
- Target cell line (e.g., HEK-293, fibroblast lines, or antigen-presenting cells like RAW 264.7).
- LNPs encapsulating reporter mRNA (e.g., GFP or Fluc).
- Cell culture medium and reagents.
- Flow cytometer, microplate reader, or bioluminescent imager.
- Cell viability assay kit (e.g., MTT or CellTiter-Glo).
- Procedure:
- Cell Seeding: Seed cells in a 96-well plate at an appropriate density and incubate for 24 hours.
- LNP Transfection: Apply a dilution series of LNPs (e.g., 10-200 ng mRNA per well) to the cells. Include untreated controls.
- Incubation: Incubate for 24-48 hours.
- Efficiency Analysis:
- Viability Analysis: Following the manufacturer's protocol, perform a cell viability assay on the same wells to calculate the percentage of viable cells relative to the untreated control.
Advanced LNP Engineering for Targeted Delivery
A key challenge in LNP-mediated reprogramming is achieving targeted delivery beyond the liver. Recent breakthroughs in LNP formulation challenge classical paradigms and enable enhanced organ specificity.
4.1 Structure-Activity Relationship (SAR) of Ionizable Lipids Research demonstrates that lipid structure critically determines LNP performance. A combinatorial library of 140 ester-core based ionizable lipids (nAcx-Cm) revealed that lipids with multiple branched chains (4-6) and a single hydrophobic tail on the branches significantly outperform their counterparts with fewer branches or double tails in mRNA delivery efficacy and endosomal escape [45] [88]. This SAR provides a design blueprint for novel lipids beyond SM-102 and ALC-0315.
4.2 Reformulating LNPs for Organ-Specific Targeting A paradigm-shifting finding is that cholesterol and phospholipid are dispensable for LNP functionality [45] [88]. The persistent liver tropism of conventional LNPs is largely driven by cholesterol, which facilitates lipoprotein coating and hepatic uptake. By formulating simplified, three-component LNPs (comprising only an ionizable lipid, a permanently cationic lipid, and a PEG-lipid), researchers achieved simultaneous mRNA accumulation and translation in the lung, with a significant reduction in off-target hepatic delivery [45]. This reformulation strategy is a powerful tool for directing reprogramming mRNA to specific extrahepatic tissues.
The diagram below illustrates the logical relationship between LNP composition, its physicochemical properties, and the resulting biological targeting.
The Scientist's Toolkit: Research Reagent Solutions
A curated list of essential materials and their functions, derived from the cited protocols and studies, is provided below to facilitate experimental setup.
Table 2: Essential Reagents for LNP Development and Testing
| Reagent / Material | Function / Application | Example Source / Citation |
|---|---|---|
| SM-102 | Ionizable lipid for balanced mRNA delivery; offers high stability and expression. | Taskcm, #TC-SM102 [84] |
| ALC-0315 | Ionizable lipid for high-efficiency delivery; induces robust immune responses. | Taskcm, #TC-ALC0315 [84] |
| DSPC | Structural phospholipid that contributes to LNP bilayer stability and integrity. | Avanti Polar Lipids, #850365P [86] |
| DOPE | Helper phospholipid that promotes membrane fusion and enhances endosomal escape. | [11] [86] |
| DMG-PEG2000 | PEG-lipid that controls particle size, improves stability, and reduces aggregation. | Avanti Polar Lipids, #880151P [86] |
| Cholesterol | Stabilizes LNP structure and promotes fusion with endosomal membranes. | Avanti Polar Lipids, #700100 [86] |
| TAS Buffer | Optimized storage buffer (Tris/Acetate/Sucrose) for enhanced LNP stability. | [86] |
| NanoAssemblr Technology | Microfluidic instrument for scalable, reproducible, and precise LNP formulation. | Precision NanoSystems [86] |
| CleanCap AG (3' OMe) | Co-transcriptional capping technology for producing high-quality, translatable mRNA. | [83] |
Conclusion
The engineering of lipid nanoparticles for mRNA reprogramming represents a paradigm shift in therapeutic development, moving from broad hepatic delivery to precise, cell-specific targeting. Key advancements in ionizable lipid design, component reformulation, and sophisticated screening methods are systematically addressing historical challenges in delivery efficiency, organ selectivity, and safety. The convergence of material science with machine learning is accelerating the rational design of next-generation LNPs, enabling transformative applications in oncology, autoimmune diseases, and regenerative medicine. Future progress hinges on deepening our understanding of structure-function relationships, validating novel formulations in clinical settings, and establishing scalable manufacturing processes. As LNP technology continues to evolve, it holds immense promise for delivering a new class of programmable, mRNA-based therapeutics that can be precisely tailored to treat a wide spectrum of human diseases.
References
- 1. An innovative iPSC Reprogramming mRNA-LNP Kit for ... [https://www.sciencedirect.com/science/article/abs/pii/S1465324925005845]
- 2. Review Lipid nanoparticles: Composition, formulation, and ... [https://www.sciencedirect.com/science/article/pii/S2329050125000580]
- 3. The Role of Four Lipid Components Of LNPs [https://www.biochempeg.com/article/362.html]
- 4. Lipid nanoparticle-based mRNA vaccines: a new frontier in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11336688/]
- 5. An ionizable lipid toolbox for RNA delivery [https://www.nature.com/articles/s41467-021-27493-0]
- 6. Cytosolic delivery of nucleic acids: The case of ionizable lipid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7995196/]
- 7. Effect of Cholesterol Content of Lipid Composition in mRNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9805379/]
- 8. Optimization of phospholipid chemistry for improved lipid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9113778/]
- 9. Investigating the functional contributions of phospholipids ... [https://www.sciencedirect.com/science/article/abs/pii/S0142961225005903]
- 10. PEGylated Lipid Nanoparticle Formulations: Immunological ... [https://pubmed.ncbi.nlm.nih.gov/37162501/]
- 11. Design Strategies for Novel Lipid Nanoparticle for mRNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12497691/]
- 12. Particles that enhance mRNA delivery could reduce ... [https://news.mit.edu/2025/particles-enhance-mrna-delivery-could-reduce-vaccine-dosage-costs-1107]
- 13. Engineering of mRNA vaccine platform with reduced lipids ... [https://www.nature.com/articles/s41467-025-63965-3]
- 14. Boosting mRNA Therapeutics: pHLIP Enhances ... [https://pubmed.ncbi.nlm.nih.gov/41137408/]
- 15. Incorporation of cellular membrane protein extracts into ... [https://pubmed.ncbi.nlm.nih.gov/40187649/]
- 16. Acuitas Therapeutics Unveils Next-Generation Lipid ... [https://crisprmedicinenews.com/press-release-service/card/acuitas-therapeutics-unveils-next-generation-lipid-nanoparticle-advancements-at-the-2025-mrna-health/]
- 17. Pharmacometric modeling of lipid nanoparticle-encapsulated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12418826/]
- 18. Cellular and biophysical barriers to lipid nanoparticle ... [https://www.nature.com/articles/s41467-025-60959-z]
- 19. Lipid nanoparticles for mRNA delivery [https://www.nature.com/articles/s41578-021-00358-0]
- 20. Recent Advances in the Lipid Nanoparticle-Mediated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10059764/]
- 21. Recent Advances in Lipid Nanoparticles and Their Safety ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11510967/]
- 22. Lipid nanoparticles for delivery of RNA therapeutics [https://pmc.ncbi.nlm.nih.gov/articles/PMC9691360/]
- 23. mRNA Delivery Technology | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/references/gibco-cell-culture-basics/transfection-basics/applications/mrna-delivery.html]
- 24. Lipid nanoparticle‐mediated RNA delivery for immune cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11628889/]
- 25. mRNA therapeutics beyond vaccines: dosing precision ... [https://pubs.rsc.org/en/content/articlehtml/2026/pm/d5pm00159e]
- 26. Clinical development of therapeutic mRNA applications [https://www.sciencedirect.com/science/article/abs/pii/S1525001625002084]
- 27. Efficacy versus immunogenicity of LNP-mediated delivery ... [https://www.sciencedirect.com/science/article/abs/pii/S0168365925006480]
- 28. CRISPR Clinical Trials: A 2025 Update [https://innovativegenomics.org/news/crispr-clinical-trials-2025/]
- 29. Press Release [https://ir.crisprtx.com/news-releases/news-release-details/crispr-therapeutics-announces-positive-phase-1-clinical-data]
- 30. Characterizing and controlling CRISPR repair outcomes in ... [https://www.nature.com/articles/s41467-025-66058-3]
- 31. Overcoming thermostability challenges in mRNA–lipid ... [https://www.nature.com/articles/s42003-024-06235-0]
- 32. Rational Design of Lipid Nanoparticles for Enhanced mRNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11855220/]
- 33. Structure-Activity Relationship of Ionizable Lipids for siRNA ... [https://pubmed.ncbi.nlm.nih.gov/40644303/]
- 34. Recent Advances in Lipid Nanoparticles for Delivery of mRNA [https://pmc.ncbi.nlm.nih.gov/articles/PMC9787894/]
- 35. Lipid nanoparticles for engineering next generation CAR T ... [https://pubs.rsc.org/en/content/articlehtml/2025/nh/d5nh00432b]
- 36. T cell-specific non-viral DNA delivery and in vivo CAR-T ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12258353/]
- 37. Liposomal lipid nanoparticles for extrahepatic delivery of ... [https://www.nature.com/articles/s41467-025-58523-w]
- 38. Replacing cholesterol and PEGylated lipids with zwitterionic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12506999/]
- 39. Delivering the Message: Translating mRNA Therapy for Liver ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12361847/]
- 40. Developing mRNA Nanomedicines with Advanced ... [https://link.springer.com/article/10.1007/s40820-025-01665-9]
- 41. Tissue-specific mRNA delivery and prime editing with ... [https://pubmed.ncbi.nlm.nih.gov/40890498/]
- 42. Peptide-modified lipid for organ-specific mRNA delivery [https://sciexplor.com/articles/bmeh.2025.0006]
- 43. Adipocyte-selective mRNA lipid nanoparticles for cell ... [https://www.sciencedirect.com/science/article/abs/pii/S0168365925007898]
- 44. Adipocyte-selective mRNA lipid nanoparticles for cell ... [https://pubmed.ncbi.nlm.nih.gov/40885402/]
- 45. Reformulating lipid nanoparticles for organ-targeted mRNA ... [https://www.nature.com/articles/s41467-024-50093-7]
- 46. Emerging Approaches for the Discovery of Lipid-Based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12473667/]
- 47. Optimization of Lipid-Nanoparticle Delivery Systems [https://www.bioprocessintl.com/vaccines/bottlenecks-in-rna-delivery-optimization-of-lipid-nanoparticle-delivery-systems]
- 48. Machine Learning Unlocks Next-Generation Lipid ... [https://inbt.jhu.edu/machine-learning-unlocks-next-generation-lipid-nanoparticles-for-safer-gene-editing/]
- 49. Machine Learning-guided Lipid Nanoparticle Design for ... [https://arxiv.org/abs/2308.01402]
- 50. Designing lipid nanoparticles using a transformer-based ... [https://www.nature.com/articles/s41565-025-01975-4]
- 51. Optimizing the targeting of lipid nanoparticles for gene therapy [https://pubs.rsc.org/en/content/articlehtml/2025/nh/d5nh00351b]
- 52. Unlocking the full therapeutic potential of lipid ... [https://www.sciopen.com/article/10.26599/NR.2025.94907422]
- 53. Low-liver-accumulation lipid nanoparticles enhance the ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-06924-2]
- 54. PEGylated lipid screening, composition optimization, and ... [https://pubs.rsc.org/en/content/articlehtml/2025/nr/d5nr00433k]
- 55. Endo/Lysosomal-Escapable Lipid Nanoparticle Platforms for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12300936/]
- 56. Endosomal escape and current obstacles in ionizable lipid ... [https://www.sciencedirect.com/science/article/pii/S0378517325011007]
- 57. Structure and Function of Cationic and Ionizable Lipids for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9812548/]
- 58. Ionizable lipid chemistry in lipid nanoparticles determines ... [https://www.sciencedirect.com/science/article/abs/pii/S0168365925006777]
- 59. Synergistic effect of nucleoside modification and ionizable ... [https://www.nature.com/articles/s41541-025-01263-1]
- 60. Ionizable lipid nanoparticles of mRNA vaccines elicit NF-κB ... [https://www.nature.com/articles/s41541-025-01124-x]
- 61. mRNA therapeutics beyond vaccines: dosing precision ... [https://pubs.rsc.org/en/content/articlehtml/2025/pm/d5pm00159e]
- 62. Progress and prospects of mRNA-based drugs in pre- ... [https://www.nature.com/articles/s41392-024-02002-z]
- 63. Hydroxychloroquine-functionalized ionizable lipids mitigate ... [https://www.sciencedirect.com/science/article/abs/pii/S016836592500879X]
- 64. Researchers Develop New LNP to Enhance The Efficacy of ... [https://www.creative-biogene.com/blog/researchers-develop-new-lnp-to-enhance-the-efficacy-of-mrna-therapy]
- 65. New nanoparticle mRNA vaccine may be cheaper and 100 ... [https://newatlas.com/medical-tech/nanoparticle-mrna-vaccine-cheaper-more-powerful/]
- 66. Reducing off-target expression of mRNA therapeutics and ... [https://www.sciencedirect.com/science/article/pii/S2329050124002183]
- 67. Artificial Intelligence-Driven Strategies for Targeted Delivery ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12389219/]
- 68. Buffer optimization of siRNA-lipid nanoparticles mitigates ... [https://www.nature.com/articles/s41467-025-63651-4]
- 69. Characterization of mRNA-LNP structural features and ... [https://www.sciencedirect.com/science/article/abs/pii/S0168365924007521]
- 70. Successful reprogramming of cellular protein production through... [https://pubmed.ncbi.nlm.nih.gov/29588418/]
- 71. Recent advances in mRNA - LNP therapeutics: immunological and... [https://jnanobiotechnology.biomedcentral.com/articles/10.1186/s12951-022-01478-7]
- 72. Biophysical Characterization of Viral and Lipid-Based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8780473/]
- 73. Standardizing a Protocol for Streamlined Synthesis and ... [https://pubmed.ncbi.nlm.nih.gov/40766677/]
- 74. Advances in mRNA - LNP lung-targeted delivery strategies [https://www.oaepublish.com/articles/microstructures.2024.172]
- 75. In Vivo Cellular Reprogramming: The Next Generation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6234007/]
- 76. Deciphering the biological fate of mRNA-LNP-based ... [https://www.sciencedirect.com/science/article/pii/S2211383525007737]
- 77. Protein corona formed on lipid nanoparticles compromises ... [https://www.nature.com/articles/s41467-025-63726-2]
- 78. Effect of mRNA-LNP components of two globally-marketed ... [https://www.nature.com/articles/s41541-023-00751-6]
- 79. Opportunities and Challenges in the Delivery of mRNA-Based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7076378/]
- 80. Versatility of LNPs across different administration routes for targeted... [https://pubs.rsc.org/en/content/articlehtml/2025/tb/d5tb00575b]
- 81. A complete guide to understanding Lipid nanoparticles (LNP) [https://insidetx.com/resources/reviews/complete-guide-to-understanding-lipid-nanoparticles-lnp/]
- 82. Quantitative analysis of lipids and nucleic acids in ... [https://www.insights.bio/cell-and-gene-therapy-insights/journal/article/3257/Quantitative-analysis-of-lipids-and-nucleic-acids-in-lipid-nanoparticles-using-mo]
- 83. Effect of mRNA-LNP components of two globally-marketed ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10567765/]
- 84. From siRNA to mRNA: Choosing the Optimal Lipid Delivery ... [https://www.taskcm.com/from-sirna-to-mrna-choosing-the-optimal-lipid-delivery-material-sm-102-alc-0315-mc3/]
- 85. Shaping the future of mRNA based drug delivery [https://www.kofo.mpg.de/1032895/2025-08-01-alc-315]
- 86. Lipid Nanoparticle Development for A Fluvid mRNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12159160/]
- 87. Novel Lipid Nanoparticles Stable and Efficient for mRNA ... [https://www.mdpi.com/1422-0067/25/3/1388]
- 88. Reformulating lipid nanoparticles for organ-targeted mRNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11226454/]