Fresh vs. Cryopreserved PBMCs for CAR-T Manufacturing: A Comprehensive 2025 Guide for Researchers
This article provides a critical analysis for researchers and drug development professionals on the use of fresh versus cryopreserved peripheral blood mononuclear cells (PBMCs) as starting material for Chimeric Antigen...
Fresh vs. Cryopreserved PBMCs for CAR-T Manufacturing: A Comprehensive 2025 Guide for Researchers
Abstract
This article provides a critical analysis for researchers and drug development professionals on the use of fresh versus cryopreserved peripheral blood mononuclear cells (PBMCs) as starting material for Chimeric Antigen Receptor T-cell (CAR-T) therapy. It explores the foundational science behind cryopreservation's impact on cell viability and phenotype, details optimized methodologies for both viral and non-viral manufacturing platforms, offers troubleshooting strategies for common challenges, and synthesizes recent clinical and pre-clinical data validating the comparability of final CAR-T products. The review concludes that cryopreserved PBMCs offer a logistically superior and functionally non-inferior alternative, enabling greater flexibility and scalability in CAR-T production without compromising therapeutic efficacy, as evidenced by 2025 clinical outcomes.
PBMC Fundamentals: How Cryopreservation Impacts Cell Biology and Viability
Peripheral Blood Mononuclear Cells (PBMCs) are a critical cellular subset that serves as the foundational starting material for manufacturing Chimeric Antigen Receptor T-cell (CAR-T) therapies. These cells are isolated from peripheral blood or leukapheresis products and consist of lymphocytes (T cells, B cells, and NK cells) and monocytes, all characterized by a single round nucleus [1]. The quality and composition of PBMCs significantly influence the success of CAR-T manufacturing, impacting everything from transduction efficiency to the final product's therapeutic potential. Within the context of CAR-T development, a central question has emerged: how does the use of cryopreserved PBMCs compare to fresh PBMCs as starting material? This guide objectively compares these two approaches, synthesizing current research data to inform decision-making by researchers, scientists, and drug development professionals.
PBMC Composition and Isolation Methods
Core Cellular Components of PBMCs
PBMCs represent a heterogeneous population of immune cells, each with distinct roles in immunity and cell therapy manufacturing.
- T Lymphocytes: The workhorses of CAR-T therapy, these cells are genetically engineered to express CARs targeting specific tumor antigens. T cells are further subdivided into helper (CD4+) and cytotoxic (CD8+) populations, both crucial for an effective anti-tumor response [2].
- B Lymphocytes: Antibody-producing cells that are often the target for CAR-T therapies in B-cell malignancies but are typically removed during T-cell enrichment for CAR-T manufacturing.
- Natural Killer (NK) Cells: Innate immune cells with cytotoxic capability that are also being explored as platforms for CAR-based therapies.
- Monocytes: Antigen-presenting cells that can differentiate into macrophages or dendritic cells, playing supporting roles in immune activation.
The initial proportion of these subsets, particularly naive and memory T cells, significantly influences the expansion potential and persistence of the final CAR-T product [2].
Techniques for PBMC Isolation
The method chosen for PBMC isolation can affect cell yield, viability, and subsequent functionality in manufacturing.
- Density Gradient Centrifugation: This widely used method separates blood components based on density using media like Ficoll-Paque. PBMCs form a distinct layer that can be collected after centrifugation. While cost-effective and capable of processing large volumes, it requires technical skill and may cause cellular stress [1].
- Magnetic-Activated Cell Sorting (MACS): This method uses antibody-coated magnetic beads to specifically target and isolate cell populations (e.g., CD4+ or CD8+ T cells). MACS offers higher specificity and purity but requires specialized equipment and reagents [1].
- Fluorescence-Activated Cell Sorting (FACS): This technique employs fluorescently labeled antibodies and flow cytometry to sort cells based on multiple surface markers simultaneously. FACS provides the highest specificity and purity but is the most technically demanding and expensive option [1].
- Microbubble Technology: An emerging gentle method that uses buoyant microbubbles to bind and float unwanted cells for removal, providing "untouched" target PBMCs with minimal stress [1].
Fresh vs. Cryopreserved PBMCs: A Comparative Analysis
The choice between fresh and cryopreserved PBMCs as starting material involves balancing manufacturing flexibility with potential impacts on cell quality. The table below summarizes key comparative findings from recent studies.
Table 1: Comparative Analysis of CAR-T Manufacturing from Fresh vs. Cryopreserved PBMCs
| Performance Metric | Fresh PBMCs | Cryopreserved PBMCs | Research Findings |
|---|---|---|---|
| Cell Viability | Higher initial viability [3] | Slightly reduced (4-9% decrease) but stable long-term [4] [3] | No significant impact on manufacturing success [4] [5] |
| T Cell Proportion | Stable baseline | Maintained post-thaw [4] | Key T-cell subsets for CAR-T production preserved after freezing [4] |
| CAR-T Expansion | Robust expansion | Comparable/slightly slower but reaches target doses [6] [7] | Final expansion yields sufficient for therapy [6] |
| Transduction Efficiency | Standard efficiency | Comparable to fresh PBMCs [8] | Successful with viral and non-viral (e.g., PiggyBac) systems [4] |
| Cell Phenotype | Reference standard | Comparable T-cell differentiation and exhaustion profiles [4] | Preserved stem-like memory populations important for persistence [4] |
| In Vitro Function | Potent cytotoxicity | Comparable tumor cell killing [4] [6] [7] | No systematic differences in cytokine release profiles [4] |
| Clinical Outcomes | Established efficacy | Similar safety, response rates, and survival [5] [8] | No significant difference in CRS, ICANS, ORR, OS, or PFS [5] |
Detailed Experimental Protocols and Data
To ensure reproducibility and provide context for the data in Table 1, this section outlines key experimental methodologies from cited studies.
Objective: To evaluate the effect of long-term cryopreservation (3 months to 2 years) on PBMC viability, phenotype, and subsequent CAR-T function.
Methods:
- PBMC Collection & Cryopreservation: PBMCs were isolated from healthy donors via density gradient centrifugation and cryopreserved in controlled-rate freezers using DMSO-containing cryoprotectant.
- Thawing and Analysis: Frozen PBMCs were thawed at designated time points, and viability was assessed using trypan blue exclusion.
- Immunophenotyping: Multicolor flow cytometry was performed using antibodies against CD3, CD4, CD8, CD45RO, and CCR7 to quantify T-cell subsets and differentiation states (naive, central memory, effector memory).
- CAR-T Generation: Thawed PBMCs were activated, transduced with a mesothelin-targeting CAR using the PiggyBac transposon system via electroporation, and expanded for 11 days.
- Functional Assays:
- Cytotoxicity: Real-time cellular analysis (RTCA) against SKOV-3 ovarian cancer cells at effector-to-target (E:T) ratios.
- Cytokine Secretion: Multiplex ELISA to measure IFN-γ, IL-2, IL-4, IL-5, IL-6, IL-10, IL-13, and TNF-α in co-culture supernatants.
Objective: To retrospectively compare the safety and efficacy of anti-CD19 CAR-T therapy in patients with Diffuse Large B-Cell Lymphoma (DLBCL) using cryopreserved versus fresh PBMCs.
Methods:
- Patient Cohort: 162 relapsed/refractory DLBCL patients were included; 136 received CAR-T from cryopreserved PBMCs, and 26 received CAR-T from fresh PBMCs.
- PBMC Processing: Cryopreserved PBMCs were frozen with a controlled-rate freezer and CryoSure-DEX40 cryoprotectant, then stored in liquid nitrogen.
- Manufacturing and Dosing: All patients received a fresh infusion of anti-CD19-4-1BB-CD3ζ CAR-T cells post-manufacture, with a target dose of 2×10⁶ cells/kg.
- Key Outcome Measures:
- Product Characteristics: Success in achieving target infusion dose, transduction efficiency.
- Clinical Efficacy: 3-month complete response (CR) and objective response rate (ORR), 1-year overall survival (OS) and progression-free survival (PFS).
- Safety Profile: Incidence and severity of cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS).
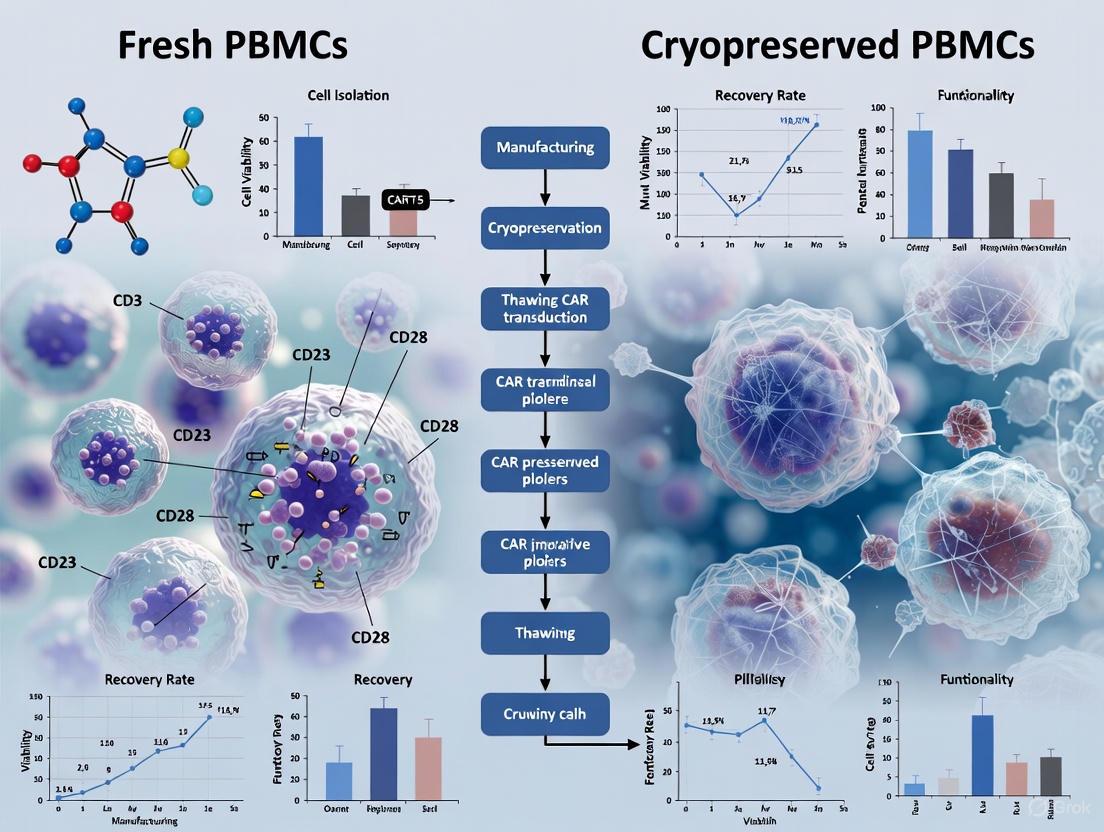
Workflow and Signaling Visualization
The following diagram illustrates the typical workflow for manufacturing CAR-T cells from fresh and cryopreserved PBMCs, highlighting key comparison points.
CAR-T Manufacturing Paths from Fresh and Cryopreserved PBMCs
The Scientist's Toolkit: Essential Research Reagents
The table below lists key reagents and materials essential for experiments comparing fresh and cryopreserved PBMCs in CAR-T manufacturing.
Table 2: Essential Reagents for PBMC and CAR-T Research
| Reagent/Material | Function | Application Example |
|---|---|---|
| Ficoll-Paque | Density gradient medium for isolating PBMCs from whole blood or leukapheresis. | Initial separation of mononuclear cells from other blood components [1]. |
| Cryoprotectant (e.g., DMSO/CS10) | Prevents ice crystal formation during freezing to maintain cell viability. | Cryopreservation of PBMCs or leukapheresis products for long-term storage [5] [3]. |
| Activation Beads (anti-CD3/CD28) | Provides stimulatory signals to activate T cells, a critical step before genetic modification. | T-cell activation prior to transduction in both fresh and cryopreserved protocols [7]. |
| Cytokines (e.g., IL-2) | Supports T-cell growth, survival, and expansion during culture. | Added to media during the CAR-T cell expansion phase [8]. |
| Viral Vectors (Lentiviral/Retroviral) | Delivers the CAR genetic construct to T cells for stable expression. | Genetic modification of activated T cells [2] [7]. |
| Transposon System (e.g., PiggyBac) | Non-viral method for integrating the CAR gene into the T-cell genome. | Electroporation-based CAR transduction, compatible with cryopreserved PBMCs [4]. |
| Flow Cytometry Antibodies | Detects surface markers (CD3, CD4, CD8) and intracellular proteins to characterize cells. | Immunophenotyping of PBMCs and CAR-T products, and detecting CAR expression [4] [7]. |
The comprehensive analysis of current research demonstrates that cryopreserved PBMCs are a viable and robust alternative to fresh PBMCs for CAR-T manufacturing. While initial post-thaw viability may be slightly lower, this does not translate to significant functional deficits in the final CAR-T product. Critical attributes including expansion potential, transduction efficiency, phenotypic profiles, in vitro cytotoxicity, and most importantly, clinical safety and efficacy, are maintained at comparable levels [4] [6] [5].
The choice between fresh and cryopreserved starting materials should be guided by practical considerations. Cryopreservation offers substantial logistical advantages, enabling manufacturing flexibility, banked starting material from healthier patients, and resilience in supply chains [4] [3]. For researchers and drug developers, this evidence supports the adoption of cryopreserved PBMCs as a standard practice, facilitating more accessible and scalable CAR-T therapy development without compromising on product quality or patient outcomes.
Cryopreservation serves as a cornerstone technology in biomedical research and cellular therapy, enabling long-term storage of vital immune cells such as peripheral blood mononuclear cells (PBMCs). In the context of chimeric antigen receptor T-cell (CAR-T) manufacturing, the decision between using fresh or cryopreserved PBMCs carries significant implications for therapeutic efficacy, manufacturing flexibility, and clinical accessibility. While fresh cells theoretically represent the most pristine biological material, logistical challenges in clinical trials and manufacturing often necessitate cryopreservation. This comprehensive analysis examines the scientific underpinnings of cryopreservation, delineates the mechanisms of cellular damage, and provides objective, data-driven comparisons between fresh and cryopreserved PBMCs for CAR-T manufacturing research.
The cryopreservation process subjects cells to extreme physical and chemical stresses, including ice crystal formation, osmotic shock, and cryoprotectant toxicity. Understanding these mechanisms is paramount for optimizing preservation protocols and maintaining cellular functionality post-thaw. This review synthesizes current research on cryopreservation outcomes, with particular emphasis on quantitative assessments of cell viability, phenotypic stability, and functional capacity—critical parameters for generating potent CAR-T products.
Fundamental Mechanisms of Cryopreservation and Cellular Damage
Principles of Cryopreservation
Cryopreservation operates on the principle that biochemical reactions and cellular metabolism effectively cease at ultra-low temperatures (typically -196°C in liquid nitrogen). Successful preservation requires navigating the liquid-to-solid phase transition of water while minimizing damage to cellular structures. The process involves three critical stages: (1) introduction of cryoprotective agents (CPAs) to suppress ice crystal formation, (2) controlled-rate freezing to promote protective dehydration, and (3) rapid thawing to minimize recrystallization damage.
During freezing, cells face two primary hazards: intracellular ice formation and "solution effects" resulting from concentration of solutes in the unfrozen fraction. Intracellular ice crystals can physically disrupt organelles and membranes, while the hypertonic extracellular environment can cause osmotic shrinkage, membrane damage, and protein denaturation. The optimal cooling rate represents a balance between these competing damage mechanisms, varying by cell type based on volume-to-surface area ratios and membrane permeability properties.
Mechanisms of Cryopreservation Damage
Ice Crystal Formation and Physical Damage: The formation and growth of ice crystals during freezing and thawing represents a primary mechanism of cellular injury. Ice crystals can physically pierce plasma membranes and intracellular organelles, leading to loss of compartmentalization and release of cytotoxic contents. For NK cells, research indicates that freezing disrupts cytolytic granules containing perforin and granzyme, compromising cytotoxic function [9].
Osmotic Stress and Volume Changes: As extracellular ice forms, solutes become concentrated in the remaining liquid, creating hypertonic conditions that draw water out of cells. This dehydration causes cell shrinkage and membrane stress. During thawing, the reverse process can cause excessive swelling and membrane rupture if not properly controlled. The large, osmotically inactive volume of NK cells demonstrates particular sensitivity to these volume changes [9].
Cryoprotectant Toxicity: While cryoprotectants like dimethyl sulfoxide (DMSO) are essential for preventing ice formation, they themselves can cause cellular damage. DMSO exerts concentration- and time-dependent toxic effects, including alterations to membrane fluidity and protein function. Exposure to cryoprotectants has been shown to reduce NK cell-induced cytotoxicity and membrane fluidity even before freezing occurs [9]. Additionally, DMSO infusion in patients can cause adverse effects ranging from nausea to cardiac arrest [9].
Oxidative Stress and Apoptosis: The freeze-thaw process generates reactive oxygen species (ROS) that can damage lipids, proteins, and DNA. This oxidative stress can trigger apoptotic pathways, leading to delayed cell death post-thaw. Studies of sperm cryopreservation have demonstrated increased DNA fragmentation and elevated apoptotic markers like Caspase-3 following freezing [10], and similar mechanisms likely affect PBMCs.
Comparative Analysis: Fresh vs. Cryopreserved PBMCs in CAR-T Manufacturing
Impact on Cell Viability and Recovery
Multiple studies have systematically evaluated the viability and recovery of PBMCs following short-term and long-term cryopreservation. The data reveal that although cryopreservation causes an immediate reduction in viability compared to fresh cells, optimized protocols can maintain viability at acceptable levels for manufacturing purposes.
Table 1: Viability and Recovery of Cryopreserved PBMCs Over Time
| Cryopreservation Duration | Viability (%) | Recovery (%) | Key Findings | Study |
|---|---|---|---|---|
| Fresh PBMCs (baseline) | 95-98% | 100% | Baseline for comparison | [4] [11] |
| 3 weeks (M0) | 90-95% | 91-95% | Minor immediate post-thaw decrease | [11] |
| 3 months (M3) | 89-94% | 89-93% | Stable performance | [4] [11] |
| 6 months (M6) | 88-93% | 87-92% | Consistent with shorter storage | [4] [11] |
| 12 months (M12) | 87-92% | 85-90% | Minimal additional decline | [4] [11] [12] |
| 24 months (M24) | 86-91% | 83-89% | Long-term viability maintained | [4] [11] |
Notably, one study examining PBMCs cryopreserved for 3.5 years still demonstrated average viability of 90.95% [4], suggesting that proper cryopreservation can maintain cell viability for extended periods. The most significant factors affecting viability include the cryopreservation medium composition, cooling rate, and thawing procedure rather than storage duration alone.
Phenotypic Stability and Cellular Composition
Maintaining appropriate cellular composition and phenotype is crucial for CAR-T manufacturing, as T-cell subsets differentially contribute to CAR-T product efficacy. Research indicates that while cryopreservation affects certain cell populations, critical T-cell subsets remain relatively stable.
Table 2: Impact of Cryopreservation on PBMC Composition and T-cell Phenotypes
| Cell Population | Impact of Cryopreservation | Functional Significance | Study |
|---|---|---|---|
| T cells (CD3+) | Proportion remains stable | Essential for CAR-T manufacturing | [4] |
| B cells | Proportion decreases | Minimal impact on CAR-T generation | [4] [13] |
| NK cells | Proportion decreases | Not used in CAR-T manufacturing | [4] |
| Naïve T cells (Tn) | No significant changes | Critical for long-term persistence | [4] |
| Central Memory T cells (Tcm) | No significant changes | Important for sustained efficacy | [4] |
| Exhaustion Markers | No systematic changes | Favorable for functional potency | [4] |
A particularly important finding for CAR-T manufacturing is that T cell differentiation states—specifically the proportions of naïve T cells (Tn) and central memory T cells (Tcm)—show no significant changes post-cryopreservation compared to fresh samples [4]. These subsets are crucial for CAR-T persistence and long-term therapeutic efficacy.
Functional Capacity for CAR-T Generation
The ultimate test of cryopreserved PBMCs is their capacity to generate functional CAR-T products comparable to those derived from fresh cells. Multiple studies have addressed this question using various genetic modification approaches.
Table 3: Functional Comparison of CAR-T Products from Fresh vs. Cryopreserved PBMCs
| Functional Parameter | Fresh PBMCs | Cryopreserved PBMCs | Significance | Study |
|---|---|---|---|---|
| Expansion Potential | Baseline | Comparable | No significant impact on manufacturing yield | [4] [5] |
| Transfection Efficiency | Baseline | Comparable | Consistent CAR expression across groups | [4] |
| Cytotoxicity | 91.02%-100% | 95.46%-98.07% | No significant functional difference | [4] |
| Cytokine Secretion | Baseline | Comparable (except IFN-γ in CAR-12M) | Minimal functional impact | [4] |
| In vivo Persistence | 21 days (median) | 21 days (median) | Equivalent persistence post-infusion | [5] |
| Clinical Response (ORR) | 69.2% | 61.9% | No statistically significant difference | [5] |
Notably, a clinical study of 162 DLBCL patients receiving anti-CD19 CAR-T therapy found no significant differences in key efficacy metrics—including overall survival, progression-free survival, and objective response rate—between those receiving CAR-T from cryopreserved versus fresh PBMCs [5]. This real-world evidence strongly supports the functional equivalence of cryopreserved starting material.
Methodological Considerations and Optimization Strategies
Cryopreservation Media and Cryoprotectants
The composition of cryopreservation media significantly influences post-thaw cell viability and function. Traditional media often incorporate fetal bovine serum (FBS) with 10% DMSO, but this formulation raises concerns about xeno-immunization, batch-to-batch variability, and potential pathogen transmission [11]. Recent systematic comparisons of commercially available serum-free media have identified viable alternatives that maintain PBMC viability and functionality comparable to FBS-based media [11].
Among serum-free options, CryoStor CS10 and NutriFreez D10 have demonstrated particularly robust performance, maintaining high viability and functionality across a 2-year storage period [11]. Media with DMSO concentrations below 7.5% generally showed significantly reduced viability, suggesting that DMSO concentration cannot be substantially reduced without compromising preservation quality [11].
Technical Protocols for Cryopreservation and Thawing
Standardized protocols are critical for reproducible cryopreservation outcomes. The following technical details represent consensus approaches derived from multiple studies:
Cryopreservation Protocol:
- Isolate PBMCs using density gradient centrifugation (e.g., Ficoll-Paque or Lymphoprep)
- Resuspend cells at optimal density (1-5×10⁶ cells/mL) in cryopreservation medium
- Use controlled-rate freezing at approximately 1°C/minute to -80°C
- Transfer to liquid nitrogen for long-term storage at -135°C to -196°C [14]
Thawing Protocol:
- Rapidly thaw cells in a 37°C water bath with gentle agitation
- Immediately dilute cryoprotectant with pre-warmed culture medium
- Add DNase (10μg/mL) to prevent cell clumping due to DNA release from damaged cells
- Wash cells to remove residual cryoprotectant [11] [12]
The cooling rate has been identified as particularly critical for specific cell types. For Natural Killer cells, studies determined an optimal cooling rate of 4-5°C/minute [9], slightly faster than the standard 1°C/minute often used for PBMCs.
Quality Assessment and Functional Validation
Rigorous quality control is essential for ensuring cryopreserved PBMCs meet manufacturing standards. Key assessment parameters include:
Viability Assessment:
- Trypan blue exclusion for immediate viability quantification [15] [12]
- Flow cytometry with viability dyes (e.g., propidium iodide) for more accurate determination [12] [14]
Functional Assays:
- Cytokine release assays (ELISA, intracellular cytokine staining) [11] [14]
- Proliferation assays (CFSE dilution) [14]
- Cytotoxicity assays against target cell lines [4]
- Fluorospot assays for antigen-specific responses [11]
Studies indicate that PBMCs with viability ≥70% are suitable for functional assays, including lymphoproliferative responses, cytokine production studies, flow cytometric analyses, and immunomagnetic cell separation [15].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Key Research Reagents for PBMC Cryopreservation and CAR-T Research
| Reagent/Category | Specific Examples | Function/Application | Considerations |
|---|---|---|---|
| Cryopreservation Media | CryoStor CS10, NutriFreez D10, Bambanker D10 | Cell protection during freezing | Serum-free options eliminate xeno-immunization risks [11] |
| Cryoprotectants | DMSO, Glycerol | Prevent ice crystal formation | DMSO concentration critical (optimum ~10%) [11] |
| Separation Media | Ficoll-Paque, Lymphoprep | PBMC isolation from whole blood | Density gradient centrifugation |
| Viability Assessment | Trypan blue, Propidium iodide, Live/Dead stains | Cell viability quantification | Flow cytometry offers highest accuracy [12] [14] |
| Cell Culture Media | RPMI-1640 with supplements | Post-thaw recovery and expansion | Often requires serum or serum alternatives |
| Characterization Antibodies | CD3, CD4, CD8, CD45RO, CCR7 | Phenotypic analysis by flow cytometry | Essential for subset quantification [4] [12] |
| Genetic Modification | PiggyBac transposon system, Lentiviral vectors | CAR introduction into T cells | Non-viral methods reducing costs [4] |
The comprehensive analysis of current research demonstrates that cryopreserved PBMCs represent a viable alternative to fresh cells for CAR-T manufacturing, with comparable performance across critical parameters including viability, phenotypic stability, expansion potential, and functional efficacy. While cryopreservation induces predictable minor deficits in immediate post-thaw viability, these limitations are offset by the significant advantages in manufacturing flexibility, logistical planning, and quality control.
The mechanistic understanding of cryopreservation damage—particularly ice crystal formation, osmotic stress, and cryoprotectant toxicity—informs continuous refinement of preservation protocols. Optimization of cryopreservation media, cooling rates, and thawing procedures has substantially improved outcomes, with current protocols capable of maintaining PBMC functionality for extended periods exceeding two years.
For CAR-T manufacturing specifically, the evidence supports strategic utilization of cryopreserved PBMCs, particularly when leveraging healthy donor cells collected at optimal health status rather than patient cells potentially compromised by disease or prior treatments. This approach enables more standardized manufacturing processes and potentially enhances therapeutic outcomes by ensuring consistent starting material quality. As cryopreservation methodologies continue to evolve, further reduction of cellular stress and functional impairment will likely expand the applications of preserved cellular products in both research and clinical settings.
The use of cryopreserved peripheral blood mononuclear cells (PBMCs) is a critical practice in biomedical research and cellular therapy, including for the manufacturing of Chimeric Antigen Receptor T-cells (CAR-T). While fresh cells are often considered the gold standard, cryopreservation offers unparalleled logistical advantages, enabling the decoupling of cell collection from manufacturing and the creation of cell banks for future use. The central question, however, remains: how does cryopreservation impact cell viability and function over time? This guide objectively analyzes experimental data on the post-thaw viability of PBMCs, comparing short-term and long-term stability up to 3.5 years, with a specific focus on implications for CAR-T manufacturing research.
Quantitative Viability and Stability Data
Numerous comparative studies have systematically quantified the viability and recovery of PBMCs after cryopreservation. The data below summarizes key findings from recent research, providing a clear comparison of performance metrics.
Table 1: Post-Thaw Viability and Recovery of Cryopreserved PBMCs
| Metric | Fresh PBMCs (Baseline) | Cryopreserved PBMCs (Post-Thaw) | Notes | Source |
|---|---|---|---|---|
| Viability (%) | ~99.0% - 99.5% | ~90.9% - 97.0% | Viability remains high post-thaw, though slightly reduced compared to fresh. | [16] |
| Viability after 3.5 years | Not Applicable | ~90.95% (Average) | Demonstrates remarkable long-term stability when properly stored. | [4] |
| Cell Recovery | Not Applicable | Comparable to baseline | Post-thaw cell counts are largely maintained, indicating good recovery. | [16] |
| T-cell Proportion (CD3+) | ~43.8% - 56.3% | ~42.0% - 51.2% | The critical T-cell population for CAR-T manufacturing is effectively preserved. | [16] |
Beyond basic viability, the retention of specific immune cell subsets and their functional characteristics is paramount.
Table 2: Phenotypic and Functional Stability of Cryopreserved PBMCs
| Cell Characteristic | Performance in Cryopreserved vs. Fresh PBMCs | Significance for CAR-T Manufacturing | Source |
|---|---|---|---|
| T-cell Phenotype Stability | No significant changes in proportions of T naïve (Tn) and T central memory (Tcm) post-cryopreservation. | Tn and Tcm cells are associated with enhanced CAR-T persistence and efficacy in vivo. | [4] |
| NK and B Cell Proportions | Decrease observed post-cryopreservation. | Less relevant for T-cell-focused manufacturing; may indicate higher sensitivity of these lineages to freeze-thaw. | [4] |
| CAR-T Cell Expansion | Comparable expansion potential from cryopreserved PBMCs. | Ensures that sufficient cell numbers for therapy can be manufactured from frozen starting material. | [4] [17] |
| CAR-T Cytotoxicity | Exhibits comparable cytotoxicity against target cancer cells (e.g., SKOV-3). | The critical tumor-killing function of the final CAR-T product is preserved. | [4] |
| Cytokine Secretion | Mostly comparable; one study noted a significant decrease in IFN-γ in 12-month samples, though cytotoxicity was unaffected. | Suggests potential for nuanced functional impacts that may require monitoring. | [4] |
Detailed Experimental Protocols
The reliability of post-thaw data is heavily dependent on standardized, optimized protocols for cryopreservation and thawing. The following methodologies are cited from the studies providing the data above.
Cryopreservation Protocol
The following protocol, derived from large-scale studies, ensures high post-thaw viability [16] [18] [19].
- Step 1: Cell Preparation. Isolate PBMCs from whole blood or leukapheresis product using density gradient centrifugation (e.g., Ficoll-Paque or CPT tubes). Centrifuge the isolated PBMCs and carefully remove the supernatant [18].
- Step 2: Cryoprotectant Formulation. Resuspend the cell pellet in a cold, serum-free cryoprotectant solution like CryoStor CS10 (containing 10% DMSO) to achieve a final concentration of 5-10 x 10^6 cells/mL [16] [19]. Using a defined, serum-free medium mitigates batch variability and safety concerns for clinical applications.
- Step 3: Controlled-Rate Freezing. Transfer the cell suspension to cryovials and immediately initiate a controlled-rate freezing process. Use an isopropanol freezing container (e.g., Mr. Frosty) placed at -80°C overnight or a programmed freezer to achieve a consistent cooling rate of approximately -1°C per minute [19].
- Step 4: Long-Term Storage. After 24 hours, transfer the cryovials to the vapor phase of liquid nitrogen (below -135°C) for long-term storage. Storage at -80°C is not recommended for long-term preservation [19].
Thawing and Viability Assessment Protocol
A gentle and rapid thawing process is critical to maximize cell recovery [18] [20].
- Step 1: Rapid Thaw. Remove vials from liquid nitrogen and immediately place them in a 37°C water bath with gentle agitation until only a small ice crystal remains.
- Step 2: Dilution and Washing. Gently transfer the cell suspension to a tube containing pre-warmed complete culture medium. Slowly add the medium drop-wise to dilute the cytotoxic DMSO. Centrifuge the cells at a gentle force (e.g., 330 x g for 10 minutes) to remove the cryoprotectant [18].
- Step 3: Viability Analysis. Resuspend the cell pellet in an appropriate buffer. Mix a sample of cells with a viability stain, such as Trypan Blue, and count using a hemocytometer or automated cell counter to determine the percentage of viable cells [18]. For immunophenotyping, cells are typically stained with fluorochrome-labeled antibodies (e.g., anti-CD3 for T-cells) and analyzed via flow cytometry [4].
The following workflow diagram summarizes the key steps from cell collection to viability assessment.
Impact on CAR-T Manufacturing Attributes
For CAR-T manufacturing, the ultimate test of cryopreserved PBMCs is the quality of the final therapeutic product. Comparative analyses reveal that CAR-T cells generated from cryopreserved PBMCs perform comparably to those from fresh cells across multiple critical quality attributes.
Table 3: CAR-T Product Attributes from Fresh vs. Cryopreserved PBMCs
| CAR-T Attribute | Fresh PBMC-Derived CAR-T | Cryopreserved PBMC-Derived CAR-T | Conclusion | Source |
|---|---|---|---|---|
| Expansion Potential | Baseline | Slight reductions in some studies, but not statistically significant. | Comparable expansion can be achieved. | [4] [17] |
| Transduction Efficiency | Baseline | Comparable. | Genetic modification is not impaired by the freeze-thaw process. | [17] |
| Cell Phenotype (at harvest) | Baseline Tn/Tcm levels. | Tn/Tcm levels show no significant difference. | Favourable phenotypes for persistence are maintained. | [4] |
| Exhaustion Markers | Baseline levels. | Consistent with fresh; no systematic increase. | Cells do not show signs of increased exhaustion. | [4] |
| In-vitro Cytotoxicity | High, baseline level. | Comparable high level of target cell killing. | Critical effector function is fully retained. | [4] [17] |
The logical relationship between starting material properties and the resulting CAR-T product quality is summarized below.
The Scientist's Toolkit: Essential Research Reagents
Successful cryopreservation and analysis depend on key reagents and materials. The following table lists essential solutions used in the featured protocols.
Table 4: Key Reagent Solutions for PBMC Cryopreservation Research
| Reagent / Material | Function / Purpose | Example Product | Protocol Notes |
|---|---|---|---|
| Cryopreservation Medium | Protects cells from ice crystal damage during freezing and thawing. Contains cryoprotectants like DMSO. | CryoStor CS10 | A defined, serum-free GMP-grade medium; reduces variability and safety risks [19]. |
| Density Gradient Medium | Isolates PBMCs from other blood components (RBCs, granulocytes) by centrifugation. | Ficoll-Paque, CPT Tubes | CPT tubes integrate blood collection and separation into a single vacuum tube [18]. |
| Cell Activation Reagents | Activates T-cells and initiates proliferation prior to genetic modification. | Anti-CD3/CD28 antibodies, IL-2 | Critical first step in CAR-T manufacturing post-thaw [17]. |
| Viability Stain | Distinguishes live cells from dead cells for counting and flow cytometry. | Trypan Blue, Propidium Iodide | Allows for accurate calculation of post-thaw viability and recovery [18]. |
| Flow Cytometry Antibodies | Identifies and characterizes specific immune cell populations (e.g., T-cells, subsets). | Anti-CD3, -CD4, -CD8, -CD45RO, -CCR7 | Used for immunophenotyping to confirm preservation of critical subsets [4]. |
The collective experimental data demonstrates that cryopreserved PBMCs are a robust and reliable starting material for CAR-T manufacturing research. While a minor decrease in immediate post-thaw viability is observed compared to fresh cells, this does not translate to a significant functional deficit. Critically, long-term cryopreservation for up to 3.5 years maintains viability at around 91%, and the resulting CAR-T products exhibit comparable expansion, phenotype, and cytotoxic functionality. The key to unlocking this performance lies in the strict adherence to standardized, optimized protocols for cryopreservation and thawing. Therefore, the use of cryopreserved PBMCs presents a valid and logistically superior alternative to fresh cells, enabling greater flexibility and resilience in the CAR-T manufacturing supply chain.
The advent of chimeric antigen receptor T-cell (CAR-T) therapy has revolutionized cancer treatment, particularly for hematologic malignancies. A critical component of CAR-T manufacturing relies on the initial quality of the source T-cells, which are typically isolated from peripheral blood mononuclear cells (PBMCs). The use of fresh versus cryopreserved PBMCs for CAR-T manufacturing represents a significant methodological consideration in both research and clinical settings, with substantial implications for logistical planning, product consistency, and therapeutic efficacy [4]. While fresh PBMCs theoretically offer unaltered cellular composition and function, cryopreserved PBMCs provide unparalleled flexibility for scheduling, manufacturing scalability, and the utilization of donor cells collected at optimal health states [4].
The central question surrounding cryopreservation revolves around its impact on phenotypic stability—specifically, whether the process alters the relative proportions and viability of critical immune cell populations, including T-cells, natural killer (NK) cells, and B-cells. Understanding these effects is paramount for researchers and drug development professionals who must make informed decisions about cell sourcing for CAR-T production. This guide objectively compares the current experimental data on phenotypic stability in fresh versus cryopreserved PBMCs, providing a scientific basis for protocol selection in CAR-T manufacturing research.
Comparative Analysis of Phenotypic Stability Post-Thaw
Viability and Cellular Composition
The immediate impact of cryopreservation on PBMCs is most evident in overall cell viability and recovery. Studies consistently report a measurable, though often moderate, decrease in viability following thawing.
Table 1: Impact of Cryopreservation on PBMC Viability and Recovery
| Storage Duration | Cell Type | Viability/Recovery | Change from Fresh | Citation |
|---|---|---|---|---|
| 6-12 months | Total PBMCs | Significantly decreased | Not quantified | [21] |
| 3 months | Total PBMCs | Relatively stable | -4.00% to -5.67% | [4] |
| 3.5 years | Total PBMCs | Relatively stable | Average 90.95% | [4] |
| 12 months | Total PBMCs (scRNA-seq) | Cell capture efficiency significantly declined | ~32% reduction | [22] |
| 12 months | NK Cells | Highly donor-dependent | 51% - 95% recovery | [23] |
Beyond general viability, the relative proportions of lymphocyte subsets demonstrate variable tolerance to the freeze-thaw process. Research indicates that T lymphocytes, particularly CD4+ T cells, are the most significantly affected, whereas monocytes, NK cells, NKT cells, and B cells appear more resilient [21]. The mechanism underlying this preferential loss of CD4+ T cells has been linked to cell death induced by elevated reactive oxygen species (ROS) [21].
T-Cell Subset Stability
The stability of T-cell subsets is of paramount importance for CAR-T manufacturing, as these cells are the direct precursors to the final therapeutic product. Investigations into both proportion and function reveal key insights.
Table 2: T-Cell Phenotype and Functional Stability After Cryopreservation
| Parameter | Cell Subtype | Finding | Significance for CAR-T | Citation |
|---|---|---|---|---|
| Proportion | Total T Cells | Remained relatively stable post-cryopreservation. | Suggests a viable T-cell source for manufacturing. | [4] |
| Proportion | CD4+ T Cells | Most significantly decreased after cryopreservation. | May impact the CD4+/CD8+ ratio in the final CAR-T product. | [21] |
| Phenotype | Tn and Tcm | No significant changes in proportions (CD45RO-CCR7+ and CD45RO+CCR7+) post-cryopreservation. | Critical for product efficacy, as Tn/Tcm are associated with enhanced persistence in vivo. | [4] |
| Function | T Cell Proliferation | Significantly affected after long-term cryopreservation. | Could influence the expansion potential during CAR-T manufacturing. | [21] |
| Function | Cytokine Secretion (IL-2) | Significantly affected after long-term cryopreservation. | Indicates potential functional impairment in activation. | [21] |
| Function | Cytokine Secretion (IFN-γ) | Significant decrease in CAR-12M vs. CAR-F in one study, but cytotoxicity was unaffected. | Highlights that functional assays may show discrepancies not affecting final product toxicity. | [4] |
| Suppressive Function | Regulatory T Cells (Treg) | Preserved after cryopreservation. | Supports the use of frozen cells for Treg-based therapies. | [24] |
A direct comparative study on generating CAR-T cells using the PiggyBac electroporation system from fresh versus cryopreserved PBMCs found that cells derived from cryopreserved PBMCs exhibited comparable expansion potential, cell phenotype, differentiation profiles, exhaustion markers, and cytotoxicity against target cancer cells to those derived from fresh PBMCs [4]. This is a critical finding for the field, as it supports the feasibility of using non-viral transfection methods with frozen starting material.
NK-Cell and B-Cell Stability
While often considered more cryo-resilient, NK cells and B cells still undergo changes that could influence their utility in research or combination therapies.
NK Cells: Although one study noted a decrease in the proportion of NK cells post-cryopreservation [4], their functionality can be more profoundly affected. Post-thaw NK cells demonstrate critical changes, including reduced viability over time in culture, decreased expression of the activating receptor NKG2D, and impaired cytotoxic activity and cytokine production [23]. This functional impairment is a significant consideration for NK cell-based immunotherapy.
B Cells: Similar to NK cells, a decrease in the proportion of B cells has been observed after cryopreservation [4]. However, the transcriptomic profiles of B cells, as part of the broader PBMC population, do not show substantial perturbation after 6 or 12 months of storage, indicating that the genetic programming remains relatively intact [22].
Detailed Experimental Protocols for Key Studies
Workflow for Assessing Long-Term Cryopreservation Impact
The following diagram outlines a comprehensive experimental workflow used to evaluate the impact of long-term cryopreservation on PBMCs and subsequent CAR-T generation.
PBMC Processing and Cryopreservation Methodology
The reliability of data comparing fresh and cryopreserved cells is highly dependent on stringent and standardized protocols. The following detailed methodology is synthesized from several studies and aligns with gold-standard recommendations, such as those from the Office of HIV/AIDS Network Coordination (HANC) [25].
Sample Collection and Isolation: Peripheral blood is collected in EDTA or heparin anticoagulant tubes [21] [25]. PBMCs are isolated using density gradient centrifugation with Ficoll-Paque [21] [22]. The HANC-SOP recommends that processing time should not exceed 8 hours to maintain optimal viability and immunogenicity [25].
Cryopreservation Protocol: Isolated PBMCs are resuspended in a cryoprotective medium, most commonly consisting of 10% Dimethyl Sulfoxide (DMSO) and 90% Fetal Bovine Serum (FBS) or human serum albumin [21] [24]. Cells are typically frozen at a controlled rate (e.g., using a CryoMed Freezer or an isopropanol-filled "Mr. Frosty" container) at -80°C before long-term transfer to liquid nitrogen (-196°C) for storage [22] [24].
Thawing and Recovery: Vials are rapidly thawed in a 37°C water bath until a small ice crystal remains [22]. The cell suspension is immediately transferred to a pre-warmed culture medium (e.g., RPMI-1640 with 10% FBS) and washed to remove DMSO [21] [22]. A critical step for functional assays is to "rest" the thawed PBMCs overnight in culture medium, often at a high density (e.g., 5-10 x 10^6 cells/mL), to recover from the freeze-thaw stress [25].
Key Analytical Assays
- Flow Cytometry: The primary tool for phenotypic analysis. It is used with fluorochrome-labeled antibodies against CD3 (T cells), CD4 (Helper T), CD8 (Cytotoxic T), CD19 (B cells), CD56/16 (NK cells), and markers for memory subsets (CD45RO, CCR7) [21] [4].
- Functional Assays:
- Proliferation: Measured using CFSE dilution, where cells are labeled with CFSE and tracked after stimulation (e.g., with anti-CD3/CD28 antibodies) [21].
- Cytokine Secretion: Using intracellular cytokine staining after stimulation with PMA/ionomycin or antigen-specific peptides, or by measuring secreted cytokines in supernatant via ELISA [21] [4].
- Cytotoxicity: Real-time cellular analysis (RTCA) or co-culture with target cells (e.g., SKOV-3) to measure specific lysis [4].
- Single-Cell RNA Sequencing (scRNA-seq): Provides unbiased analysis of transcriptomic profiles and cellular heterogeneity, identifying differentially activated pathways and subtle changes in gene expression [21] [22].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents and Their Applications in PBMC/CAR-T Research
| Reagent / Solution | Primary Function | Example Use in Protocol |
|---|---|---|
| Ficoll-Paque / Lymphoprep | Density gradient medium for isolating PBMCs from whole blood. | Centrifugation to separate PBMC layer from other blood components. |
| DMSO (Dimethyl Sulfoxide) | Cryoprotective agent; prevents ice crystal formation to protect cells during freezing. | Used at 5-10% in cryopreservation medium, often with FBS. |
| Fetal Bovine Serum (FBS) | Component of cryopreservation and culture media; provides proteins and growth factors. | 90% FBS + 10% DMSO is a common cryopreservation cocktail. |
| Recombinant Human IL-2 | Cytokine used to support T-cell and NK cell survival and proliferation in culture. | Added to culture media during CAR-T expansion and post-thaw recovery. |
| Anti-CD3/CD28 Antibodies | Synthetic activation stimuli mimicking TCR engagement; used for T-cell activation and expansion. | Bead-bound or soluble antibodies used to activate T-cells prior to genetic modification. |
| CFSE (Carboxyfluorescein succinimidyl ester) | Fluorescent cell staining dye that dilutes with each cell division; used to track proliferation. | Labeling cells before stimulation to monitor division cycles via flow cytometry. |
| PMA/Ionomycin | Pharmacological agents that strongly activate T-cells, bypassing the TCR; used for intracellular cytokine staining. | Stimulating cells in the presence of Brefeldin A to detect cytokine production potential. |
| PiggyBac Transposon System | Non-viral vector for integrating large DNA sequences (e.g., CAR transgene) into the host cell genome. | Electroporation of CAR transposon and transposase plasmids into activated T-cells. |
The collective experimental data indicates that cryopreservation of PBMCs is a viable strategy for CAR-T manufacturing research, though it is not without its specific effects. The most consistent findings across studies are a moderate reduction in overall viability and a selective loss of CD4+ T cells, potentially mediated by ROS-induced apoptosis. However, the critical T-cell phenotypes for CAR-T efficacy—naïve and central memory T-cells—remain proportionally stable after thawing.
Furthermore, functional comparisons demonstrate that CAR-T cells generated from cryopreserved PBMCs can exhibit comparable expansion, cytotoxicity, and phenotypic profiles to those derived from fresh PBMCs, particularly when optimized protocols for the non-viral PiggyBac transposon system are employed [4]. The preservation of Treg suppressive function post-thaw further supports the use of cryopreservation in other cell therapy contexts [24].
For researchers, the decision to use fresh or cryopreserved PBMCs should be guided by the specific requirements of the study. If the utmost preservation of all original immune cell subsets, particularly CD4+ T cells, is critical, fresh cells may be preferable. However, for most CAR-T manufacturing applications, where logistical flexibility and the use of donor cells from a healthy state are paramount, cryopreserved PBMCs represent a robust and reliable alternative, provided that standardized, optimized protocols for freezing, thawing, and recovery are rigorously followed.
The quality of the starting T-cell population is a critical determinant of success in Chimeric Antigen Receptor T-cell (CAR-T) therapy, with naïve (Tn) and central memory (Tcm) T cells being particularly vital due to their enhanced expansion potential, persistence, and antitumor efficacy [4] [2]. A central question in CAR-T manufacturing research is whether cryopreserved peripheral blood mononuclear cells (PBMCs) can serve as a reliable alternative to their fresh counterparts without compromising these essential T-cell subsets. This guide provides a comparative analysis of fresh and cryopreserved PBMCs, synthesizing experimental data on the recovery, phenotypic stability, and functional capacity of Tn and Tcm cells to inform strategic decisions in therapeutic cell manufacturing.
Quantitative Comparison of T-cell Subsets in Fresh vs. Cryopreserved PBMCs
The following tables consolidate key quantitative findings from comparative studies, offering a clear overview of the impact of cryopreservation on critical T-cell attributes.
Table 1: Impact of Cryopreservation on PBMC Viability and T-cell Proportion Stability
| Parameter | Fresh PBMCs | Cryopreserved PBMCs | Significance | Source |
|---|---|---|---|---|
| Cell Viability | Baseline | 4.00% to 5.67% decrease post-thaw | Significant, but minor absolute decrease | [4] |
| T-cell Proportion (CD3+) | Stable baseline | Remained relatively stable | No significant change; CAR-T preparation unaffected | [4] |
| Naïve T-cell (Tn) Proportion | Stable baseline | No significant change post-cryopreservation | Phenotype preserved | [4] |
| Central Memory (Tcm) Proportion | Stable baseline | No significant change post-cryopreservation | Phenotype preserved | [4] |
| NK and B-cell Proportions | Stable baseline | Decreased post-cryopreservation | Sensitive to hypothermic conditions | [4] |
Table 2: Functional Profile of CAR-T Cells Generated from Fresh vs. Cryopreserved PBMCs
| Functional Attribute | CAR-T from Fresh PBMCs | CAR-T from Cryopreserved PBMCs | Significance | Source |
|---|---|---|---|---|
| Expansion Potential | Baseline | Comparable | No significant impact observed | [4] |
| Cytotoxicity | 91.02% - 100.00% (at E:T 4:1) | 95.46% - 98.07% (at E:T 4:1) | Comparable anti-tumor activity | [4] |
| T-cell Exhaustion Markers | Baseline | Consistent levels | No significant increase | [4] |
| Cytokine Secretion (IFN-γ) | Baseline | Significant decrease in CAR-12M | Cytotoxic function remained unaffected | [4] |
| Clinical Outcomes (NHL) | Baseline (Fresh CAR-T product) | Similar antitumor efficacy and safety | Cryopreservation is a suitable formulation | [26] |
Experimental Protocols for Comparative Analysis
To ensure the reliability and reproducibility of data comparing fresh and cryopreserved cells, standardized experimental protocols are essential. Below are detailed methodologies for key assays cited in this guide.
Protocol 1: Multicolor Flow Cytometry for Immunophenotyping
This protocol is used to quantify T-cell subsets and assess phenotypic markers [4] [27].
- Sample Preparation: Isolate PBMCs from whole blood via density gradient centrifugation (e.g., using Ficoll). Split the sample for immediate analysis (fresh) and for cryopreservation.
- Cryopreservation Procedure: Resuspend PBMC pellets in freezing media (e.g., 10% DMSO, 40% FCS, 50% RPMI-1640). Aliquot cells into cryovials and freeze using a controlled-rate freezer (e.g., "Mr. Frosty" at -80°C) before transferring to long-term storage at -150°C [4] [27].
- Thawing and Staining: Rapidly thaw cryopreserved vials in a 37°C water bath. Wash cells in PBS with 2% FCS to remove DMSO. Allow cells to rest for 1 hour at room temperature before staining.
- Antibody Staining: Stain cell suspensions with fluorochrome-conjugated antibodies against surface markers (e.g., CD3, CD4, CD8, CD45RO, CCR7) for 20 minutes at room temperature in the dark [27]. For intracellular cytokine staining, stimulate cells first, then permeabilize and stain.
- Data Acquisition and Analysis: Acquire data using a flow cytometer (e.g., CytoFLEX LX). Analyze using software such as CytExpert or FlowJo. Gate on lymphocytes, exclude doublets and dead cells, then identify T-cell subsets (e.g., Tn as CD45RO-CCR7+; Tcm as CD45RO+CCR7+) [4] [27].
Protocol 2: Real-Time Cellular Analysis (RTCA) for Cytotoxicity
This functional assay measures the ability of generated CAR-T cells to kill target tumor cells [4].
- Effector and Target Cell Preparation: Generate CAR-T cells from both fresh and cryopreserved PBMCs via activation and genetic modification. Culture target tumor cells (e.g., SKOV-3 for ovarian cancer).
- Assay Setup: Seed target cells into specialized RTCA plates. After the target cells adhere, add CAR-T effector cells at specified Effector-to-Target (E:T) ratios (e.g., 4:1 and 2:1). Include control wells with target cells only and non-transduced T cells (Mock-T).
- Monitoring and Analysis: Place the plate in the RTCA instrument, which continuously monitors electrical impedance. Cell death or detachment causes impedance changes, which are recorded and analyzed by the instrument's software. Cytotoxicity is calculated based on these kinetic measurements [4].
Protocol 3: Cytokine Release Assay
This protocol assesses the functional secretory profile of T cells upon activation [4].
- Stimulation: Co-culture CAR-T cells with target cells or stimulate with agents like PMA/ionomycin in the presence of a protein transport inhibitor.
- Supernatant Collection: After a defined incubation period (e.g., 24 hours), centrifuge the culture plates and collect the supernatant.
- Cytokine Measurement: Use a multiplex bead-based immunoassay (e.g., Luminex) or ELISA to quantify the concentration of multiple cytokines (e.g., IFN-γ, IL-2, IL-6, TNF-α) in the supernatant, according to the manufacturer's instructions [4].
Workflow and Pathway Visualization
The following diagrams illustrate the core comparative experimental workflow and the T-cell differentiation pathway relevant to assessing subset quality.
Comparative Analysis Workflow
T-cell Differentiation and Memory Subset Relationships
The Scientist's Toolkit: Key Research Reagents and Materials
Table 3: Essential Reagents and Tools for PBMC and T-cell Studies
| Item | Function/Application | Example Specifics |
|---|---|---|
| Cryopreservation Media | Protects cells from ice crystal damage during freeze-thaw. | 10% DMSO in Normosol-R [28] or clinical-grade CS10 [16]. |
| Controlled-Rate Freezer | Ensures reproducible, optimal cooling rates for high viability. | "Mr. Frosty" or planar Kryo 560; critical for process standardization [4] [16]. |
| Magnetic Cell Separation Kits | Isolation of specific T-cell populations (e.g., CD4+, CD8+) from PBMCs. | EasySep Human T Cell Isolation Kit; CD3/CD28 beads for activation [29] [2]. |
| Flow Cytometry Antibody Panels | Immunophenotyping of T-cell subsets (Tn, Tcm, Tem) and exhaustion markers. | Antibodies against CD3, CD4, CD8, CD45RO, CCR7, PD-1, LAG-3 [4] [27] [29]. |
| Cell Culture Media & Cytokines | Ex vivo T-cell activation and expansion for CAR-T manufacturing. | RPMI-1640 supplemented with IL-2 (activation) and IL-7/IL-15 (expansion) [29] [2]. |
| Real-Time Cell Analyzer (RTCA) | Label-free, dynamic assessment of CAR-T mediated cytotoxicity. | Measures impedance changes in co-culture with target cells [4]. |
From Thaw to Transduction: Optimized Protocols for Cryopreserved PBMCs
Best Practices for Thawing and Washing Cryopreserved PBMCs to Maximize Recovery
In CAR-T manufacturing research, the choice between using fresh versus cryopreserved PBMCs carries significant implications for production logistics and experimental flexibility. While fresh leukapheresis offers minimal pre-processing manipulation, cryopreserved PBMCs provide substantial advantages for scalable, distributed manufacturing models by decoupling cell collection from processing timelines [3]. The critical determinant in this comparison often hinges on post-thaw recovery and functionality, which are profoundly influenced by thawing and washing techniques. Optimal thawing protocols directly impact cell viability, recovery rates, and downstream functionality—key concerns for researchers and drug development professionals aiming to maximize the value of precious cellular resources [30] [25]. This guide systematically compares methodological approaches to PBMC thawing, providing evidence-based recommendations to ensure consistent, high-quality results for CAR-T and other immunology research applications.
Thawing Methodologies: Experimental Data and Comparisons
Temperature Conditions: Warm vs. Cold Processing
The temperature of both thawed cells and washing media significantly impacts PBMC recovery and functionality. Research systematically testing permutations of common thawing practices has revealed clear performance differences between "warm" and "cold" processing approaches.
Table 1: Comparison of Warm vs. Cold Thawing Processing on PBMC Viability and Functionality
| Processing Condition | Cell Viability | CD8+ T-cell Functionality | CD4+ T-cell Functionality | Key Findings |
|---|---|---|---|---|
| Warm Processing (37°C media added slowly to 37°C cells) | High (Median 96.6% when washed immediately) [30] | Preserved (No significant reduction) [30] | Preserved (Significantly higher than cold processing) [30] | Superior for maintaining viability and CD4+ T-cell responses in ELISPOT assays |
| Cold Processing (Ice-chilled media rapidly added to ice-cold cells) | Significantly reduced (p=0.002) [30] | Moderate reduction (1.98-fold diminution) [30] | Severely impaired (12.05-fold diminution) [30] | Rapid addition of cold media particularly detrimental; slow addition partially mitigates viability loss |
The experimental data reveals that adding ice-chilled media rapidly to ice-cold cells represents a high-risk practice that strongly reduces viable PBMC recovery [30]. While slow addition of cold media can partially overcome this drop in viability, warm processing conditions consistently yield superior results for both cell recovery and functionality, particularly for CD4+ T-cell responses [30].
DMSO Exposure and Washing Strategies
The cryoprotectant DMSO presents a paradoxical consideration in thawing protocols: while prolonged exposure is potentially toxic, its rapid elimination must be balanced against procedural cell loss.
Table 2: Impact of Washing Steps and DMSO Exposure on PBMC Quality
| Parameter | Experimental Findings | Impact on PBMC Quality |
|---|---|---|
| Number of Washes | Two washes significantly increased viability compared to a single wash (p<0.05) [30] | Enhanced cell viability and significantly improved CD4+ T-cell functionality in ELISPOT assays |
| DMSO Exposure at 37°C | Minimal viability loss after 30 minutes (94.2% vs. 96.6% at 0 minutes); moderate decline after 60 minutes (93.0%) [30] | Surprisingly low risk; permits batch-thawing in high-throughput environments without significant viability loss |
| Post-Thaw Resting | Recommended before functional assays; allows recovery from activation and stress-induced genes [25] [12] | Improves response to stimuli and restores more native transcriptional state |
These findings demonstrate that while DMSO removal is important, the methodical execution of washing is more critical than speed alone. The unexpected tolerance of PBMCs to DMSO exposure at 37°C for up to 30 minutes provides valuable flexibility for processing multiple samples in batch workflows [30].
Detailed Experimental Protocols
Optimal Thawing and Washing Protocol
Based on comparative analysis, the following protocol represents current best practices for thawing cryopreserved PBMCs:
Preparation: Warm complete culture medium (e.g., RPMI-1640 with 10% FBS) to 37°C. Pre-warm centrifuge to room temperature.
Thawing: Remove cryovial from liquid nitrogen and immediately place in a 37°C water bath. Gently swirl until only a small ice crystal remains (approximately 1-2 minutes) [30] [19].
Initial Dilution: Transfer the cell suspension to a 15mL conical tube. Slowly add 10mL of pre-warmed medium dropwise over 60 seconds while gently swirling the tube [30].
Washing: Centrifuge at 300-500 × g for 10 minutes at room temperature [24] [19]. Carefully decant supernatant without disturbing the pellet.
Second Wash: Resuspend cells in 10mL warm medium and repeat centrifugation. Note that the two-step washing process significantly enhances viability and functionality compared to single wash protocols [30].
Final Resuspension: Resuspend cell pellet in appropriate volume of culture medium for counting and downstream applications.
For high-throughput environments where immediate washing of all samples is impractical, research indicates that thawed PBMCs in cryovials can be maintained at 37°C for up to 30 minutes before washing commences without significant viability loss [30].
Suboptimal Thawing Protocol (For Comparison)
To illustrate practices that compromise recovery, the following protocol should be avoided:
- Thaw cryovial in 37°C water bath until last ice crystals disappear.
- Immediately add ice-cold media to ice-cold cells rapidly (in <10 seconds).
- Perform only a single wash step before proceeding to downstream applications.
This approach has been experimentally shown to significantly reduce viability and impair CD4+ T-cell functionality [30].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for PBMC Thawing and Processing
| Reagent/Consumable | Function | Implementation Notes |
|---|---|---|
| DMSO (10%) | Cryoprotectant in freezing medium | Use clinical-grade (e.g., CryoStor CS10) for manufacturing; toxicity increases with prolonged room temperature exposure [19] |
| Fetal Bovine Serum (FBS) | Provides proteins that protect cells during freezing/thawing | Use 90% FBS with 10% DMSO for research; not recommended for clinical applications due to variability [19] |
| Water Bath | Ensures consistent, rapid thawing | Set to 37°C; verify temperature calibration regularly [30] |
| Pre-warmed Culture Medium | Dilutes and removes DMSO while maintaining cellular homeostasis | RPMI-1640 with 10% FBS and HEPES buffer recommended; maintain at 37°C [30] [12] |
| Isopropanol Freezing Container | Provides controlled-rate freezing | Mr. Frosty or Corning CoolCell; ensures -1°C/minute freezing rate for optimal viability [19] |
Thawing Process Workflow
The following diagram illustrates the critical decision points in the PBMC thawing process and their impacts on cell quality:
Implications for CAR-T Manufacturing Research
The methodological comparisons presented here carry particular significance for CAR-T manufacturing research, where the quality of starting materials directly influences production success. Recent multi-platform comparative studies demonstrate that cryopreserved leukapheresis products can achieve ≥90% post-thaw viability with recovery and phenotypic profiles comparable to peripheral blood mononuclear cells (PBMCs) [3]. Importantly, cryopreserved starting materials have shown higher lymphocyte proportions (66.59% vs. 52.20% in PBMCs), potentially enhancing CAR-T manufacturing potential [3].
While some studies note that fresh CAR-T infusion products may exhibit increased in vitro anti-tumor reactivity, cryopreserved products maintain high anti-tumor potency and specificity without compromising clinical outcomes [8]. This supports the feasibility of cryopreserved PBMCs as starting material, particularly when optimal thawing practices are implemented.
The logistical advantages of cryopreserved materials are substantial for distributed manufacturing models, enabling decoupling from fresh material logistics and improving supply chain resilience [3]. When researchers employ the evidence-based thawing methods outlined herein, cryopreserved PBMCs represent a viable, and in some aspects advantageous, alternative to fresh cells for CAR-T manufacturing research.
The comparative analysis of PBMC thawing methodologies reveals that temperature control during processing, methodical washing techniques, and judicious DMSO management collectively determine post-thaw cell quality. The experimental data consistently demonstrates that warm processing conditions with slow addition of pre-warmed media followed by two wash steps yields superior viability and functionality preservation. For CAR-T manufacturing research specifically, these optimized protocols enable researchers to leverage the practical advantages of cryopreserved materials without compromising cellular integrity or function. By implementing these evidence-based practices, research and drug development professionals can maximize the value of their cellular resources while maintaining the flexibility required for complex therapeutic development pipelines.
In the field of Chimeric Antigen Receptor T-cell (CAR-T) manufacturing, the debate between using fresh versus cryopreserved peripheral blood mononuclear cells (PBMCs) as starting material is crucial for process optimization. While fresh leukapheresis has been the traditional standard, its time-sensitive viability decay and complex logistics present significant challenges for scalable manufacturing [16] [3]. Cryopreserved leukapheresis offers a promising alternative by decoupling manufacturing from fresh material logistics, thereby enhancing supply chain resilience [16]. This guide provides a comprehensive comparison of performance metrics between fresh and cryopreserved materials, with focused experimental data on optimizing centrifugation procedures, cryoprotectant proportions like CS10, and implementing automated closed systems.
Comprehensive Performance Comparison: Fresh vs. Cryopreserved Starting Materials
Extensive research has systematically evaluated the impact of cryopreservation on key quality attributes of starting materials and the resulting CAR-T products. The data below summarizes critical comparative findings.
Table 1: Performance Comparison of Fresh vs. Cryopreserved Starting Materials
| Parameter | Fresh Leukapheresis | Cryopreserved Leukapheresis | Cryopreserved PBMCs | Significance |
|---|---|---|---|---|
| Initial Viability (%) | 99.0 - 99.5% [16] | 90.9 - 97.0% [16] | Varies with medium [31] | Slightly lower initial viability in cryo, but functionally recovers. |
| Lymphocyte Proportion | 68.68 ± 1.78% [3] | 66.59 ± 2.64% [3] | 52.20 ± 9.29% [3] | Cryo-leukapheresis retains significantly higher lymphocytes than cryo-PBMCs. |
| Post-Thaw Viability Benchmark | Not Applicable | ≥ 90% [16] | ≥ 90% (in CS10/NutriFreez D10) [31] | Achievable with optimized process. |
| T-cell Profile (CD3+ %) | 43.82 - 56.31% [16] | 42.01 - 51.21% [16] | Information Missing | Minimal loss of T-cells during processing and cryopreservation. |
| CAR-T Manufacturing Compatibility | Yes (Reference) | Yes (Non-viral, Lentiviral, Fast CAR-T platforms) [16] | Yes [8] | Comparable in expansion, phenotype, and cytotoxicity. |
| Clinical Outcome Correlation | Established | No negative impact observed [8] | No negative impact observed [8] | Use of frozen products is a viable option. |
Table 2: Comparison of Final CAR-T Cell Products from Different Starting Materials
| CAR-T Product Quality Attribute | Manufactured from Fresh Leukapheresis | Manufactured from Cryopreserved Leukapheresis | Manufactured from Cryopreserved PBMCs |
|---|---|---|---|
| Cell Viability & Expansion | Reference Standard | Comparable [16] | Sufficient for treatment [8] |
| Cell Phenotype | Reference Standard | Comparable [16] | Phenotypic differences observed (e.g., higher TIM-3 in fresh) [8] |
| CAR+ Cell Proportion | Reference Standard | Comparable [16] | Similar transduction efficacy [8] |
| In Vitro Cytotoxicity | Reference Standard | Comparable [16] | High, though potentially lower than fresh [8] |
Optimized Protocols for Cryopreservation
Centrifugation and Pre-processing Optimization
The primary challenge in cryopreserving leukapheresis products, as opposed to purified PBMCs, is managing non-target cellular impurities like red blood cells and platelets. An optimized centrifugation strategy is critical to mitigate their impact on post-thaw T-cell viability and final CAR-T product quality [16].
Key Experimental Protocol Steps [16]:
- Initial Processing: Leukapheresis products are processed through a closed automated system.
- Centrifugation Parameters: A centrifugation-based strategy is systematically implemented to remove erythrocytes and platelets. The specific g-force and time should be determined to maximize impurity removal while minimizing white blood cell loss.
- Cell Concentration Adjustment: The median cell concentration is progressively reduced from 5.09-9.71 × 10^7 cells/ml at initial leukapheresis to 4.06-5.12 × 10^7 cells/ml pre-cryopreservation through this process.
- Quality Check: Post-centrifugation viability typically ranges from 94.0 - 96.15%, and the CD3+ T lymphocyte proportion is maintained between 41.19 - 56.45%, indicating no significant T-cell loss [16].
Cryoprotectant Proportion and Formulation
The choice and concentration of cryoprotectant are vital for post-thaw recovery. DMSO is the most common cryoprotectant, but its concentration and the overall formulation must be optimized to balance cell protection and cytotoxicity.
Experimental Data on Cryoprotectants:
- DMSO Concentration: Studies on PBMCs show that media containing 10% DMSO, such as CryoStor CS10 and NutriFreez D10, consistently maintain high cell viability and functionality over long-term storage (up to 2 years), performing comparably to traditional FBS-based media [31]. Media with DMSO concentrations below 7.5% showed significant viability loss and were not recommended for long-term storage [31].
- Standardized Formulation for Leukapheresis: The optimized protocol uses a clinical-grade cryoprotectant like CS10 (10% DMSO). The final formulation ensures a DMSO concentration of 7.5% - 10% in the cryomedium, accounting for residual volume from the leukapheresis product [16].
- Cell Concentration and Volume: The target cell concentration for freezing is 5–8 × 10^7 cells/ml, with a formulation volume of 20 ml per bag, ensuring at least 1 × 10^9 cells per bag as a Critical Quality Attribute (CQA) [16].
The following diagram illustrates the optimized workflow for processing and cryopreserving leukapheresis material:
Automated and Closed System Integration
Automation is a key enabler for standardizing the cryopreservation process, reducing variability, and improving efficiency.
Implementation Protocol [16]:
- System: Utilize a closed-system automated platform for cell formulation.
- Process Efficiency: This optimization reduces processing times to a range of 43–108 minutes.
- Time-Sensitive Freezing: The interval from cryoprotectant addition to the initiation of controlled-rate freezing is strictly limited to ≤ 120 minutes. This is validated using systems like the Thermo Profile 4 to prevent ice crystal formation and ensure post-thaw viability meets the ≥ 90% benchmark [16].
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Reagents and Materials for Optimized Cryopreservation
| Item | Function/Description | Example Products/Catalogs |
|---|---|---|
| Cryoprotectant (10% DMSO) | Serum-free freezing medium protecting cells during freeze-thaw cycle. | CryoStor CS10 [16] [31], NutriFreez D10 [31] |
| Closed Automated System | Automated platform for standardized cell processing and formulation. | Platform used in study [16] |
| Controlled-Rate Freezer | Equipment ensuring standardized, reproducible freezing curve. | Thermo Profile 4 system [16] |
| Lymphocyte Separation Medium | Density gradient medium for PBMC isolation from whole blood or leukapheresis. | Lymphoprep [31], Ficoll-Hypaque [8] |
| Cell Viability Assays | Methods for assessing post-thaw cell health and recovery. | Trypan Blue Exclusion, Annexin V/PI Staining [32] [31] |
| Cell Phenotyping Assays | Flow cytometry-based analysis of cell surface markers (e.g., CD3+, CD4+, CD8+). | Antibodies for CD3, CD4, CD8, etc. [16] [8] |
The experimental data and protocols presented demonstrate that cryopreserved leukapheresis, when processed through an optimized protocol involving meticulous centrifugation, standardized cryoprotectant formulation with CS10, and integrated automated closed systems, constitutes a universal and robust raw material for CAR-T manufacturing. It preserves critical quality attributes—T-cell fitness and CAR functionality—without compromising consistency compared to fresh starting material [16]. This approach effectively decouples manufacturing from the logistical constraints of fresh materials, thereby significantly improving supply chain resilience. Future work should focus on broader protocol standardization and large-scale clinical validation to fully integrate this optimized process into mainstream CAR-T therapeutic frameworks [16].
The manufacturing of Chimeric Antigen Receptor T (CAR-T) cells is a critical step in determining the efficacy and safety of this revolutionary cancer immunotherapy. Two prominent methods for introducing the CAR gene into T cells are viral transduction using lentiviral (LV) vectors and non-viral transfection via the PiggyBac (PB) transposon system combined with electroporation. The choice between these platforms dictates specific protocol adaptations, particularly when considering the source of T cells—specifically, the use of fresh versus cryopreserved Peripheral Blood Mononuclear Cells (PBMCs). This guide objectively compares the performance of these two manufacturing platforms, providing structured experimental data and protocols to inform researchers and drug development professionals working within the context of CAR-T manufacturing optimization.
Platform Performance and Phenotypic Comparison
Direct comparisons of CAR-T cells manufactured via the LV and PB platforms reveal distinct phenotypic and functional attributes. The data below summarizes key differences observed in cytokine secretion, cell phenotypes, and transcriptional profiles.
Table 1: Comparative Phenotype and Functional Output of LV- vs. PB-derived CAR-T Cells
| Parameter | Lentiviral (LV) CAR-T Cells | PiggyBac (PB) CAR-T Cells | References |
|---|---|---|---|
| Cytokine Secretion (upon activation) | Increased IL-10 | Increased TNF-α and IFN-γ; singular expression of IL-9 | [33] [34] |
| CAR Expression Profile | More uniform, moderate CAR expression | A small fraction of cells exhibits very high CAR expression | [34] |
| Memory Phenotype | More pronounced memory phenotype (CD45RO+ CCR7+) | Comparable cytotoxic subsets, but different memory composition | [34] |
| Transcriptomic Signature | Distinct from PB CAR-T cells | Vast disparities vs. LV; greater upregulation of cytokines, chemokines, and their receptors | [34] |
| In Vitro Cytotoxicity | Strong and specific cytotoxicity | Faster initial cytotoxicity; similarly strong specific lysis | [33] [34] |
| In Vivo Anti-tumor Efficacy | Robust anti-tumor activity, enhancing survival in models | Similar strong anti-tumor activity; high doses critically effective | [33] [34] |
Detailed Experimental Protocols
To ensure reproducibility and provide a clear understanding of the technical requirements for each platform, the following section outlines the core methodologies as described in the literature.
Protocol 1: Manufacturing CD19-Targeting CAR-T Cells via PiggyBac Electroporation
This protocol is adapted from studies that successfully generated CAR-T cells from both fresh and cryopreserved PBMCs [33] [4] [34].
- PBMC Source and Preparation: Use fresh or cryopreserved PBMCs. Thaw cryopreserved PBMCs and confirm viability remains high (e.g., >90%) [4] [5].
- T Cell Activation: Resuspend PBMCs in AIM-V medium and activate them with anti-CD3/CD28-coated beads at a bead-to-cell ratio of 1:1. Include IL-2 (e.g., 100 UI) in the culture medium [33] [34].
- Electroporation:
- Timing: Perform electroporation 2-3 days post-activation [34].
- DNA Preparation: For every 1 × 10^6 primary T cells, prepare a plasmid mix containing 1.4 μg of the CD19-targeting CAR transposon vector and 0.7 μg of the Super PiggyBac transposase vector [34].
- Electroporation Buffer: Resuspend the cell-DNA mixture in a proprietary electroporation buffer.
- Instrument Parameters: Using a Celetrix electroporator, apply a pulse of 500 V for 20 ms [34]. Other studies using Lonza devices have employed conditions of 320 V for 20 ms [4].
- Post-Transfection Culture: Immediately transfer electroporated cells to pre-warmed, antibiotic-free culture medium (e.g., X-VIVO 15 or AIM-V). Continue culture with IL-2 supplementation for expansion, typically harvesting around day 11-13 [33] [4].
Protocol 2: Manufacturing CD19-Targeting CAR-T Cells via Lentiviral Transduction
This protocol details the common steps for LV-based CAR-T generation, which has also been applied to cryopreserved PBMCs [35] [34] [5].
- PBMC Source and Preparation: Use fresh or cryopreserved PBMCs. Thaw cryopreserved cells and allow for recovery in culture [5].
- T Cell Activation: Suspend PBMCs at a concentration of 1 × 10^6 cells/mL and activate with anti-CD3/CD28-coated beads at a 1:1 ratio for 24 hours [34].
- Lentiviral Transduction:
- Viral Vector: Use a second-generation lentiviral system with a transfer vector containing the CD19-CAR construct under the EF-1α promoter. The packaging plasmid pCMV-dR8.2 dvpr has been reported to yield higher titers than psPAX2 [35].
- Transduction Enhancer: Use protamine sulfate (at 8 µg/mL) or similar reagents to enhance transduction efficiency. Note that polybrene (Pb) may induce cellular senescence in some primary cells and is less desirable [36].
- Multiplicity of Infection (MOI): Infect activated T cells with lentivirus at an MOI of 3.0 [34].
- Concentration Method: Concentrating the virus with ultracentrifugation can produce higher viral titers (>5 × 10^5 IFU/mL) [35].
- Post-Transduction Culture: Refresh the medium 24 hours post-transduction. Continue culturing the cells in medium supplemented with IL-2 for expansion, with a typical culture duration similar to the PB method [33].
The Scientist's Toolkit: Essential Research Reagents
The table below lists key reagents and their functions critical for the successful execution of the protocols described above.
Table 2: Key Reagent Solutions for CAR-T Cell Manufacturing
| Reagent / Material | Function in Protocol | Examples / Notes |
|---|---|---|
| Anti-CD3/CD28 Beads | T cell activation and expansion | Mimics natural TCR co-stimulation; critical for initial T-cell activation in both platforms. |
| Recombinant IL-2 | T cell growth and survival cytokine | Maintains T cell proliferation and viability during the in vitro culture phase. |
| PiggyBac Transposon System | Non-viral gene delivery | Consists of a transposon vector (carrying the CAR) and a transposase vector (e.g., hyperactive bz-hyPBase). |
| Lentiviral Vector System | Viral gene delivery | Second-generation system (packaging, envelope, and transfer plasmids) is common for its titer and safety profile [35]. |
| Electroporator Device & Buffer | Physical delivery of DNA into cells | Devices such as Lonza/VPA-1002 or Celetrix CTX-1500A; specific buffers are often proprietary. |
| Transduction Enhancer | Improves viral vector entry | Protamine Sulfate is preferred over polybrene for some primary cells due to lower toxicity [36]. |
| Cryoprotectant | Preservation of PBMCs | CryoSure-DEX40 or similar, used with controlled-rate freezing for long-term PBMC storage [5]. |
Process Workflow and Decision Logic
The following diagram illustrates the key procedural steps and decision points for generating CAR-T cells using either the Lentiviral or PiggyBac platform, highlighting the parallel processes and critical differences.
Performance with Cryopreserved PBMCs
A critical consideration in protocol adaptation is the use of cryopreserved PBMCs, which offer logistical flexibility. Research indicates that long-term cryopreservation maintains PBMC viability and T-cell proportion relatively stable, making them a feasible source for CAR-T manufacturing [4]. The table below summarizes the comparative impact of the cell source on the final CAR-T product for both platforms.
Table 3: Impact of Cryopreserved PBMCs on CAR-T Cell Manufacturing and Quality
| Aspect | Lentiviral Platform | PiggyBac Platform | References |
|---|---|---|---|
| Feasibility | Well-established for cryopreserved PBMCs | Demonstrated as feasible with optimized protocols | [4] [5] |
| Cell Viability Post-Thaw | High viability maintained | Slight significant decrease post-thaw, but viability recovers | [4] |
| CAR-T Expansion | Comparable expansion from fresh PBMCs | Slight reduction in proliferation, but not significant | [4] |
| Phenotype (Tcm/Tn) | Stable T cell phenotypes post-cryopreservation | No significant changes in Tn and Tcm proportions vs. fresh | [4] |
| Cytotoxic Function | No significant difference in cytotoxicity vs. fresh | Comparable cytotoxicity against target cells (e.g., SKOV-3) | [4] [5] |
| Clinical Outcomes | Similar overall survival, response rates, and adverse events | Data shows comparable in vivo anti-tumor efficacy | [5] |
Both lentiviral transduction and PiggyBac electroporation are robust platforms for generating potent CAR-T cells. The choice between them involves a strategic trade-off: the lentiviral system offers a well-characterized, standardized process with a pronounced memory phenotype, while the PiggyBac system provides a cost-effective, non-viral alternative capable of achieving very high CAR expression in a subset of cells, which may be critical for dose-dependent efficacy. Critically, both platforms are compatible with cryopreserved PBMCs, enabling flexible manufacturing workflows without compromising key quality attributes of the final CAR-T product. This compatibility is pivotal for broadening the accessibility and practicality of CAR-T therapy.
In the rapidly advancing field of CAR-T cell therapy, the use of cryopreserved peripheral blood mononuclear cells (PBMCs) presents a transformative opportunity to overcome logistical hurdles and standardize manufacturing protocols. However, a significant challenge persists: initial expansion lag post-thaw. This phenomenon, characterized by delayed proliferation and reduced viability in the early culture phases, can impact manufacturing timelines and potentially affect product characteristics [4]. The metabolic and signaling stress induced by cryopreservation and recovery processes necessitates optimized culture conditions to support cell recovery and rapid expansion [37]. This guide objectively compares current strategies involving culture media formulation and cytokine supplementation to address this critical bottleneck, providing researchers with data-driven insights for protocol development.
Comparative Analysis of Media Formulations
The foundation of successful post-thaw recovery lies in selecting an appropriate basal medium. Different media formulations offer distinct nutrient compositions, osmotic balances, and support systems that significantly influence initial cell recovery and expansion.
Performance of Commercial Media
Table 1: Comparison of Commercial Media for Cryopreserved PBMC Culture
| Medium | Viability Support | Phenotype Maintenance | Key Characteristics | Reported Performance |
|---|---|---|---|---|
| RPMI 1640 | High | Superior | Lower glucose/amino acids, higher phosphate; optimal for innate immune gene expression [38]. | Superior viability maintenance and antigen-presenting cell activation [38]. |
| IMDM | High | Moderate | Rich formulation; additional vitamins and HEPES buffer [38]. | Supports good IFN-γ production from T cells [38]. |
| DMEM | Moderate | Lower | High glucose; may lead to excessive glycolysis and differentiation [37]. | Lower cell viability over extended culture [38]. |
| AIM-V | Good (Serum-Free) | Good (Serum-Free) | Chemically defined, animal-component free; supports B cell IgG responses [39]. | Viable alternative to human serum-containing media [39]. |
| X-VIVO 15 | Good (Serum-Free) | Good (Serum-Free) | Serum-free, optimized for human immune cells [39]. | Supports T and B cell proliferation and function [39]. |
Advanced Media Optimization Strategies
Emerging approaches move beyond standard formulations. Bayesian Optimization (BO) has been employed to develop optimized media blends, systematically combining commercial media like DMEM, AR5, XVIVO, and RPMI to maximize PBMC viability ex vivo [40]. This machine learning-guided method can identify non-intuitive formulations that outperform any single standard medium, achieving high cell viability with significantly reduced experimental burden compared to traditional Design of Experiments (DoE) approaches [40].
Cytokine Supplementation Strategies
Cytokines provide critical signals that direct T cell metabolism and differentiation. Strategic supplementation is essential to counteract the initial lag following cryopreservation.
Metabolic Modulation via Cytokine Selection
Table 2: Cytokine Effects on T Cell Metabolism and Differentiation
| Cytokine / Inhibitor | Primary Signaling Pathway | Metabolic Effect | Impact on Differentiation | Considerations |
|---|---|---|---|---|
| IL-2 | JAK-STAT | Promotes glycolysis and rapid proliferation [37]. | Drives effector differentiation; can limit memory populations [37]. | Standard cytokine; may exacerbate differentiation. |
| IL-7/IL-15 | JAK-STAT | Promotes oxidative phosphorylation (OXPHOS) and fatty acid oxidation (FAO) [37]. | Enhances stem-like/memory phenotypes (TSCM, TCM) [37]. | Favors persistence-associated phenotypes. |
| PI3K/Akt/mTOR Inhibitors | PI3K-AKT-mTOR | Indirectly reduces glycolysis [37]. | Increases Tn and TCM proportions [37]. | Requires precise dosing and timing. |
| 2-Deoxy-D-glucose (2-DG) | Glycolysis Inhibitor | Directly inhibits hexokinase, reducing glycolytic flux [37]. | Promotes memory cell formation and lymphoid homing [37]. | Can impair overall expansion if overused. |
Impact of Cryopreservation on Cytokine Responses
Understanding the altered responsiveness of cryopreserved cells is crucial. Single-cell analyses reveal that cryopreservation distinctly alters cytokine secretion profiles compared to fresh PBMCs. Following stimulation, cryopreserved samples show lower frequencies of cells secreting IL-6 (with LPS stimulation) and IFN-γ (with CD3/CD28 stimulation) [41]. Notably, a disconnect between cytokine expression and secretion was observed for TNF-α, where expression increased post-cryopreservation but secretion remained stable [41]. These findings underscore the need for tailored cytokine supplementation protocols specifically designed for cryopreserved starting materials.
Experimental Protocols for Method Validation
To ensure reliable comparison and implementation of these strategies, standardized experimental protocols are essential.
Protocol: Media Performance Assessment
- Cell Source: Cryopreserved PBMCs from healthy donors (stored for 3-24 months) [4].
- Thawing Procedure: Rapid thaw at 37°C, immediate transfer to pre-warmed complete medium, centrifugation to remove cryoprotectant (e.g., DMSO) [16].
- Culture Conditions: Seed cells at 0.5-1 × 10^6 cells/mL in test media (RPMI, DMEM, IMDEM, AIM-V, X-VIVO) with consistent cytokine supplementation (e.g., IL-2) [38] [39].
- Viability Assessment: Measure post-thaw viability (e.g., via Trypan Blue exclusion) and subsequently at 24, 48, and 72 hours using flow cytometry with viability dyes (e.g., Zombie Green) [41] [39].
- Phenotypic Analysis: At 72 hours, stain cells with fluorochrome-labeled antibodies against CD45RO and CCR7 to quantify naïve (TN), central memory (TCM), and effector memory (TEM) populations via flow cytometry [4].
Protocol: Cytokine Supplementation Timing
- Stimulation: Activate thawed PBMCs with anti-CD3/CD28 beads.
- Experimental Groups:
- Group 1: Standard IL-2 (100 IU/mL) throughout culture.
- Group 2: IL-7 (10 ng/mL) + IL-15 (10 ng/mL) throughout culture.
- Group 3: IL-2 for first 48 hours, then switch to IL-7/IL-15.
- Group 4: IL-7/IL-15 with late addition of IL-2.
- Metabolic Assessment: On day 3-4, analyze metabolic state by measuring glucose uptake and mitochondrial mass/function using fluorescent probes (e.g., MitoTracker) via flow cytometry [37].
- Functional Readout: On day 7-10, evaluate expansion (fold-increase), perform intracellular staining for exhaustion markers (e.g., PD-1, TIM-3), and conduct cytotoxicity assays against target tumor cells (e.g., SKOV-3) [4].
Visualizing Workflows and Pathways
The following diagrams illustrate the core experimental workflow and the underlying metabolic pathways targeted by optimization strategies.
Experimental Workflow for Optimization
Metabolic Pathways in T Cell Activation
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Optimizing Cryopreserved PBMC Culture
| Reagent Category | Specific Examples | Primary Function | Considerations for Cryopreserved Cells |
|---|---|---|---|
| Basal Media | RPMI 1640, AIM-V, X-VIVO 15 [38] [39] | Provides essential nutrients, vitamins, and buffer system. | RPMI shows superior performance for mixed PBMC populations; serum-free options (AIM-V) reduce background in Ig assays [39]. |
| Cryopreservation Media | CS10 (10% DMSO) [16] | Protects cells from ice crystal formation during freeze-thaw. | Standardized, clinical-grade cryoprotectant ensures consistent post-thaw viability ≥90% [16]. |
| T-cell Activation | Anti-CD3/CD28 Beads | Mimics antigen presentation, provides Signal 1 and 2 for T cell activation. | Critical for initiating proliferation; required after thawing to overcome initial lag. |
| Cytokines (Effector-Promoting) | Recombinant Human IL-2 | Drives rapid proliferation and expansion through metabolic shift to glycolysis [37]. | Can promote terminal differentiation; timing and concentration are critical. |
| Cytokines (Memory-Promoting) | Recombinant Human IL-7, IL-15 | Supports memory cell survival and promotes OXPHOS/FAO metabolic state [37]. | Favors persistence-associated phenotypes; ideal for later culture stages. |
| Metabolic Modulators | 2-Deoxy-D-glucose (2-DG) [37] | Inhibits hexokinase, reducing glycolytic flux. | Can uncouple expansion from differentiation, promoting memory cells; requires careful titration [37]. |
| Viability & Phenotyping Assays | Trypan Blue, Flow Cytometry with Viability Dyes (Zombie Green), Antibodies (CD45RO, CCR7) [4] [39] | Quantifies live cell recovery and T cell differentiation states. | Essential for tracking recovery post-thaw and validating strategy effectiveness. |
Addressing the initial expansion lag in cryopreserved PBMCs is a critical step toward robust and scalable CAR-T manufacturing. The experimental data and comparisons presented in this guide demonstrate that no single medium or cytokine protocol is universally superior. Rather, the optimal strategy likely involves a carefully balanced approach: a supportive basal medium like RPMI 1640 or a machine learning-optimized blend, combined with cytokine supplementation that is strategically timed to first support recovery and then steer differentiation toward favorable, persistent phenotypes. As the field moves towards allogeneic and off-the-shelf therapies, further refinement of these foundational culture parameters will be essential for unlocking the full potential of cryopreserved starting materials, ultimately enhancing the consistency, accessibility, and efficacy of cell-based immunotherapies.
For researchers and drug development professionals in cell therapy, the choice of starting material is a critical strategic decision. While cryopreserved Peripheral Blood Mononuclear Cells (PBMCs) have been a long-standing resource for research and manufacturing, cryopreserved leukapheresis is emerging as a powerful, scalable alternative that preserves a more complete immune cell repertoire. This guide provides an objective, data-driven comparison of these two materials, focusing on their performance in the context of CAR-T manufacturing research. The evidence indicates that cryopreserved leukapheresis offers significant advantages in cell yield, lymphocyte recovery, and manufacturing consistency, without compromising key CAR-T cell quality attributes, presenting a compelling case for its adoption in scalable production workflows.
Cryopreserved PBMCs are isolated mononuclear cells (lymphocytes and monocytes) that have been separated from other blood components via density gradient centrifugation (e.g., Ficoll) before cryopreservation. This process, while effective for isolation, can lead to selective cell loss, particularly of monocytes [3].
Cryopreserved leukapheresis involves the cryopreservation of the entire leukapheresis product—a concentrated collection of white blood cells obtained via apheresis. This method avoids the initial density gradient step, thereby preserving a broader spectrum of leukocytes, including granulocytes and higher numbers of monocytes and lymphocytes, which are critical for robust T-cell activation [3] [42].
Table: Fundamental Characteristics of Cryopreserved PBMCs and Leukapheresis
| Characteristic | Cryopreserved PBMCs | Cryopreserved Leukapheresis |
|---|---|---|
| Source Material | Whole blood or leukapheresis | Peripheral blood via apheresis |
| Primary Composition | Mononuclear cells (T cells, B cells, NK cells, monocytes) | All leukocytes (including granulocytes); rich in lymphocytes |
| Pre-processing | Requires density gradient centrifugation | Can be cryopreserved with minimal processing |
| Typical Use Cases | Early-stage R&D, proof-of-concept studies, immunological assays | Scalable process development, clinical manufacturing, allogeneic therapy development |
Comparative Performance Data
Cell Yield, Viability, and Composition
A direct comparison of cell composition reveals a statistically significant advantage for leukapheresis in lymphocyte recovery, which is directly relevant for T-cell therapies.
Table: Quantitative Comparison of Post-Thaw Cell Attributes
| Performance Metric | Cryopreserved PBMCs | Cryopreserved Leukapheresis | Significance/Notes |
|---|---|---|---|
| Post-thaw Viability | ~90.95% (after 3.5 years) [4] | ≥ 90% [3] | Both maintain high viability with optimized protocols |
| Lymphocyte Proportion | 52.20% ± 9.29 [3] | 66.59% ± 2.64 [3] | p < 0.05; Leukapheresis provides a larger T-cell pool |
| T-cell Proportion Stability | Remains stable post-thaw [4] | Minimal variation (e.g., 42.01–51.21% post-thaw) [3] | No significant loss of T cells during processing |
| Total Cell Yield per Unit | ~1 billion (from buffy coat) [42] | Up to 8.5 billion [42] | Leukapheresis yields are substantially higher |
Functional Performance in CAR-T Manufacturing
Critical comparative studies demonstrate that cryopreserved leukapheresis performs equivalently to its fresh counterpart and cryopreserved PBMCs in generating functional CAR-T products.
Table: CAR-T Manufacturing Outcomes from Different Starting Materials
| CAR-T Attribute | CAR-T from Cryopreserved PBMCs | CAR-T from Cryopreserved Leukapheresis |
|---|---|---|
| Cell Expansion | Comparable to fresh PBMCs; slight non-significant reduction possible [4] | Comparable to fresh leukapheresis in multiple platforms [3] |
| Transduction Efficiency | Unaffected by cryopreservation [8] | Comparable to fresh leukapheresis [3] |
| Phenotype (CD4+/CD8+) | Consistent proportions [4] | Comparable to fresh leukapheresis [3] |
| T-cell Exhaustion Markers | No significant negative impact from cryopreservation [4] | Comparable profiles to fresh material [3] |
| In-vitro Cytotoxicity | High and specific anti-tumor potency retained [8] | Comparable to fresh leukapheresis [3] |
| Cytokine Secretion | Mostly consistent; occasional variation (e.g., IFN-γ) with no functional impact [4] | Comparable profiles across key cytokines [3] |
A multi-platform comparative study concluded that in non-viral CAR-T, lentiviral CAR-T, and Fast CAR-T platforms, cryopreserved and fresh leukapheresis were comparable in cell viability, expansion, phenotype, CAR+ cell proportion, and cytotoxicity [3]. Similarly, a study on CD19 CAR-T cells found that the use of frozen PBMCs as starting material was a viable option, as frozen products still exhibited high in vitro potency and cryopreservation did not seem to affect the clinical outcome [8].
Detailed Experimental Protocols
To ensure the validity of the data presented, this section outlines the key methodologies used in the cited comparative studies.
Standardized Cryopreservation Protocol for Leukapheresis
The following optimized protocol for cryopreserving leukapheresis products was established through a closed automated system [3]:
- Centrifugation: A centrifugation-based strategy is first implemented to remove non-cellular impurities like residual red blood cells and platelets, which can impact post-thaw T-cell viability and recovery.
- Formulation: The leukocytes are resuspended in a clinical-grade cryoprotectant, CS10 (10% DMSO). The cell concentration is targeted at ~5 x 10⁷ cells/ml.
- Fill and Freeze: The formulated product is aliquoted into cryobags (e.g., 20 ml/bag). The interval from cryoprotectant addition to the initiation of controlled-rate freezing is strictly limited to ≤ 120 minutes.
- Controlled-Rate Freezing: Cryopreservation is performed using a validated controlled-rate freezer (e.g., Thermo Profile 4 system) to prevent ice crystal formation and ensure post-thaw viability ≥ 90%.
CAR-T Manufacturing and Analytical Workflow
The general workflow for comparing starting materials, as used in multiple studies, is summarized below. This process evaluates the critical quality attributes of the final CAR-T product.
Diagram: Experimental Workflow for Comparative Studies
Key Analytical Assays for Comparability
The following assays are critical for a head-to-head functional comparison:
- Flow Cytometry: For immunophenotyping of starting materials (T-cell, B-cell, NK-cell, monocyte subsets) and final CAR-T products (CAR+ percentage, CD4/CD8 ratio, differentiation markers like CCR7/CD45RO, and exhaustion markers like PD-1, TIM-3, LAG-3) [3] [43] [8].
- In Vitro Cytotoxicity Assays: To measure the specific lysis of target tumor cells. Real-time cellular analysis (RTCA) is a robust method used in recent studies [4].
- Cytokine Release Assays: Using ELISA or multiplex bead arrays to quantify secretion of key cytokines (e.g., IFN-γ, IL-2, TNF-α) upon antigen-specific stimulation [8] [4].
- Proliferation and Expansion Tracking: Monitoring cell counts and fold expansion over the culture period to assess growth potential [3] [4].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful transition and implementation rely on specific, high-quality materials and platforms.
Table: Key Reagents and Platforms for Working with Cryopreserved Leukapheresis
| Item | Function/Role | Examples / Key Specifications |
|---|---|---|
| Leukapheresis Product | The raw starting material, rich in leukocytes. | Collected via apheresis systems (e.g., Terumo Spectra Optia); requires appropriate anticoagulant (e.g., ACD-A) [42] [44]. |
| Cryoprotectant | Prevents ice crystal formation and osmotic shock during freeze-thaw. | Clinical-grade DMSO-based solutions (e.g., CS10) at final concentrations of 5-10% [3] [45]. |
| Controlled-Rate Freezer | Ensures reproducible and optimal cooling rates for high viability post-thaw. | Critical for protocol standardization; e.g., Thermo Profile series [3]. |
| Closed System Automation | For processing and formulation; reduces contamination risk and improves consistency. | Enables standardized, automated centrifugation and formulation steps [3]. |
| Cell Separation Reagents | For isolation of specific cell types post-thaw, if needed. | Immunomagnetic beads for T-cell selection (e.g., CD4/CD8 beads) [4]. |
| CAR Transduction System | For genetic modification of T cells. | Both viral (Lentiviral, Retroviral) and non-viral (PiggyBac electroporation) systems have been validated with cryopreserved leukapheresis [3] [4]. |
The collective evidence strongly supports the transition from cryopreserved PBMCs to cryopreserved leukapheresis for scalable CAR-T manufacturing research. The key advantages of leukapheresis include its superior cell yield, higher lymphocyte proportion, and reduced processing-induced cell loss, which provide a more robust and consistent starting material.
For researchers planning this transition, the following steps are recommended:
- Protocol Development: Establish a standardized, optimized cryopreservation protocol for leukapheresis, paying close attention to centrifugation for impurity removal, cryoprotectant formulation, and controlled-rate freezing.
- Comparative Validation: Conduct a head-to-head study using the experimental workflow and analytical assays outlined in this guide to benchmark performance against your current PBMC-based process.
- Logistical Planning: Leverage the decoupling of cell collection from manufacturing timelines that cryopreservation enables, thereby enhancing supply chain resilience for both autologous and allogeneic therapy development [3].
Cryopreserved leukapheresis represents a critical evolution in cell therapy raw materials, directly addressing the need for scalable, distributed, and robust manufacturing models. Its adoption is a significant step toward improving the consistency, accessibility, and ultimately, the success of advanced cell therapies.
Solving Common Challenges in Manufacturing with Frozen Starting Material
The transition from using fresh to cryopreserved peripheral blood mononuclear cells (PBMCs) in chimeric antigen receptor T-cell (CAR-T) manufacturing represents a pivotal advancement in addressing critical logistical challenges in cellular therapy. While cryopreserved PBMCs offer enhanced flexibility for clinical trial scheduling and multi-site coordination [11], this approach introduces a fundamental technical challenge: maintaining high post-thaw viability and functionality comparable to fresh cells. The process of cryopreservation exposes cells to extreme conditions that can compromise cellular integrity through ice crystal formation, osmotic stress, and molecular damage [46]. This comprehensive analysis examines two fundamental technical parameters—cryopreservation media composition and controlled-rate freezing protocols—that significantly influence post-thaw outcomes for PBMCs destined for CAR-T manufacturing.
Comparative Analysis of Cryopreservation Media Formulations
The selection of cryopreservation media profoundly impacts cell viability and functionality post-thaw. Traditional laboratory-formulated media often incorporate fetal bovine serum (FBS) with dimethyl sulfoxide (DMSO) as a cryoprotectant. However, this formulation presents significant limitations for clinical applications, including batch-to-batch variability, risk of pathogen transmission, and potential introduction of xenogenic antigens that may trigger unwanted immune responses during cell culture [19] [11]. These concerns have driven the development of serum-free, chemically defined alternatives that maintain cell viability while addressing the regulatory challenges associated with clinical-grade manufacturing.
Quantitative Comparison of Media Performance
Table 1: Comparative analysis of cryopreservation media formulations and their performance characteristics
| Media Formulation | Key Components | DMSO Concentration | Post-Thaw Viability* | Functional Preservation | Recommended Applications |
|---|---|---|---|---|---|
| FBS + DMSO [19] | 90% FBS + 10% DMSO | 10% | 84-86% [4] | Maintains T-cell functionality [11] | Research settings |
| CryoStor CS10 [19] [11] | Serum-free, defined formulation | 10% | 94.3-97.9% [47] | High functionality in cytokine release assays [11] [47] | Clinical CAR-T manufacturing |
| NutriFreez D10 [11] | Serum-free, protein-free | 10% | Comparable to CryoStor CS10 [11] | Maintains immune response [11] | Clinical & research biobanking |
| Synth-a-Freeze [48] | Defined, protein-free, no antibiotics/hormones | 10% | Not specifically reported for PBMCs | Optimized for primary cells | Stem cells & primary cells |
| Recovery Cell Culture Freezing Medium [48] | Contains FBS | 10% | ~25% higher vs. other media in cell lines | Improved recovery for cell lines | Cell lines (CHO, HEK293) |
Note: Viability percentages represent ranges across multiple studies and donor samples
Recent comprehensive research has systematically evaluated these media formulations over an extended 2-year period to assess long-term stability [11]. This study demonstrated that PBMCs cryopreserved in CryoStor CS10 and NutriFreez D10 maintained high viability and functionality comparable to the traditional FBS-based reference medium across all timepoints. Notably, media formulations with DMSO concentrations below 7.5% showed significantly reduced viability and were eliminated from consideration after initial assessments, highlighting the critical role of sufficient cryoprotectant concentration [11].
Impact on CAR-T Manufacturing Outcomes
The choice of cryopreservation media directly influences critical quality attributes of the resulting CAR-T products. Studies comparing CAR-T cells generated from cryopreserved versus fresh PBMCs have revealed no significant differences in expansion potential, cell phenotype, differentiation profiles, exhaustion markers, or cytotoxicity against tumor cell lines [4]. Importantly, clinical outcomes for patients receiving anti-CD19 CAR-T therapy demonstrated no statistically significant differences in complete response rates (46.2% vs. 45.5%), overall survival (75.4% vs. 64.1% at 1 year), or progression-free survival (52.1% vs. 44.5% at 1 year) between cryopreserved and fresh PBMC-derived products [5].
The Critical Role of Controlled-Rate Freezing
While cryopreservation media provide essential biochemical protection, the physical process of freezing equally determines post-thaw viability. Uncontrolled freezing results in rapid temperature decline that promotes intracellular ice crystal formation, which mechanically damages cellular membranes and organelles [46]. Controlled-rate freezing implements a gradual, standardized cooling profile (approximately -1°C per minute) that allows water to migrate out of cells before freezing, thereby minimizing intracellular ice formation and reducing osmotic stress [19].
Technical Implementation of Controlled-Freezing Protocols
Table 2: Comparison of controlled-rate freezing methods for PBMC cryopreservation
| Freezing Method | Cooling Rate | Technical Principle | Viability Outcomes | Implementation Considerations |
|---|---|---|---|---|
| Isopropanol Containers (e.g., CoolCell) [19] | ~-1°C/minute | Passive cooling using isopropanol as thermal buffer | High viability maintained [19] [11] | Low cost, simple operation, suitable for multi-site studies |
| Programmable Controlled-Rate Freezers [19] | Precisely controlled -1°C/minute | Active cooling with liquid nitrogen or mechanical refrigeration | Optimal preservation of sensitive cell types [46] | Higher cost, requires validation, ideal for centralized facilities |
| Passive Freezing at -80°C | Variable, often rapid | Direct placement in ultra-low freezer | Reduced and inconsistent viability [46] | Not recommended for clinical applications |
The consistent implementation of controlled-rate freezing protocols is particularly crucial for CAR-T manufacturing, where preserving the naive and central memory T-cell subsets directly correlates with in vivo persistence and therapeutic efficacy [4]. Research has demonstrated that cryopreserved PBMCs maintained stable proportions of Tn (CD45RO-CCR7+) and Tcm (CD45RO+CCR7+) populations compared to fresh samples, enabling the generation of CAR-T products with enhanced potential for long-term persistence [4].
Integrated Workflow for Optimal PBMC Cryopreservation
The following experimental workflow diagram illustrates the integrated process for cryopreserving PBMCs using optimized media and freezing conditions for CAR-T manufacturing:
Detailed Protocol for PBMC Cryopreservation
Option 1: Cryopreservation with Serum-Free Media (Recommended for Clinical Applications) [19]
- Cell Preparation: Ensure PBMCs are in a single-cell suspension. Centrifuge at 300 × g for 10 minutes to pellet cells.
- Media Exchange: Carefully remove supernatant, leaving a small amount to avoid disturbing the cell pellet. Resuspend pellet by gentle flicking.
- Cryomedium Addition: Add cold (2-8°C) CryoStor CS10 to achieve final concentration of 0.5-10 × 10⁶ cells/mL. Mix thoroughly and transfer to cryovials.
- Pre-freeze Incubation: Incubate cells at 2-8°C for 10 minutes to allow cryoprotectant equilibration.
- Controlled-Rate Freezing: Place cryovials in isopropanol freezing container (e.g., CoolCell) and transfer to -80°C freezer overnight, or use programmable freezer at -1°C/minute.
- Long-Term Storage: Transfer vials to vapor phase liquid nitrogen (-135°C to -196°C) for long-term storage. Avoid long-term storage at -80°C.
Option 2: Cryopreservation with Traditional FBS/DMSO Media [19]
- Solution Preparation: Prepare 20% DMSO in FBS. Keep on ice (note: 100% DMSO will crystallize on ice).
- Cell Preparation: Centrifuge PBMCs at 300 × g for 10 minutes. Remove supernatant and resuspend in cold FBS to concentration of 1-20 × 10⁶ cells/mL. Keep on ice.
- Mixing: Gently mix cells with 20% DMSO in FBS at 1:1 ratio (final concentration: 10% DMSO, 90% FBS).
- Rapid Processing: Quickly transfer 1 mL aliquots to cryovials and immediately place in isopropanol freezing container.
- Freezing and Storage: Place container in -80°C freezer overnight, then transfer to vapor phase liquid nitrogen.
Essential Reagents and Equipment for PBMC Cryopreservation
Table 3: Research reagent solutions for optimized PBMC cryopreservation workflows
| Reagent/Equipment | Specific Examples | Function | Considerations |
|---|---|---|---|
| Serum-Free Cryomedium | CryoStor CS10 [19] [47] | Cell protection during freeze-thaw | cGMP-manufactured, defined formulation |
| Traditional Cryomedium | 90% FBS + 10% DMSO [19] | Cell cryopreservation | Batch variability, xenogenic risk |
| Controlled-Rate Freezing Device | CoolCell [19] | Maintains -1°C/minute cooling | Compatible with standard cryovials |
| Cryogenic Storage Vials | Corning Cryogenic Vials [19] | Secure sample containment | Ensure sterility and proper labeling |
| Liquid Nitrogen Storage | Vapor phase nitrogen tanks [19] | Long-term storage below -135°C | Prevents cross-contamination |
| Post-Thaw Assessment | Flow cytometry, viability stains [4] [11] | Quality control | Assess recovery and functionality |
The integration of optimized cryopreservation media formulations with controlled-rate freezing protocols effectively mitigates the challenge of reduced post-thaw viability in PBMCs for CAR-T manufacturing. Serum-free, cGMP-manufactured media such as CryoStor CS10 and NutriFreez D10 provide superior protection compared to traditional FBS-containing formulations while addressing regulatory concerns for clinical applications. When combined with consistent controlled-rate freezing at approximately -1°C per minute, these solutions enable the generation of CAR-T products from cryopreserved PBMCs with phenotypic characteristics, expansion potential, and cytotoxic functionality comparable to those derived from fresh cells [4] [5]. This technical advancement significantly enhances the flexibility and accessibility of CAR-T manufacturing, supporting broader implementation of this transformative immunotherapy.
Overcoming Slower Early-Stage Proliferation in Non-Viral Electroporation Systems
Non-viral electroporation has emerged as a promising alternative to viral vectors for Chimeric Antigen Receptor T-cell (CAR-T) manufacturing, offering benefits such as reduced immunogenicity, lower cost, and larger cargo capacity. However, its adoption has been hampered by challenges, including notably slower early-stage T-cell proliferation post-transfection. This guide objectively compares the performance of CAR-T products generated from fresh versus cryopreserved Peripheral Blood Mononuclear Cells (PBMCs) using non-viral electroporation systems, with a focus on identifying the root causes of delayed proliferation and presenting optimized solutions supported by experimental data.
The manufacturing of CAR-T cells typically involves genetically engineering a patient's or donor's T cells to express a chimeric antigen receptor that redirects them to recognize and kill tumor cells. While viral vectors have been the traditional mainstay for gene delivery, non-viral methods, particularly electroporation, are gaining prominence due to their favorable safety profile and cost-effectiveness [49] [50].
Electroporation uses electrical pulses to create transient pores in the cell membrane, allowing plasmid DNA or other nucleic acids to enter the cell. Despite its advantages, a significant drawback is the slower early-stage proliferation of T cells observed after the procedure. This sluggish expansion can prolong manufacturing time, potentially compromise product quality, and has been a barrier to the widespread clinical application of non-viral methods [4] [50]. This performance gap is often analyzed in the context of a critical starting material decision: the use of fresh versus cryopreserved PBMCs.
Comparative Performance: Fresh vs. Cryopreserved PBMCs in Electroporation
A pivotal 2025 study provided a direct, quantitative comparison of CAR-T cells generated from fresh and cryopreserved PBMCs using the PiggyBac transposon system delivered via electroporation [4]. The key performance metrics are summarized in the table below.
Table 1: Comparative Performance of CAR-T Cells Derived from Fresh vs. Cryopreserved PBMCs via Electroporation
| Performance Metric | Fresh PBMCs | Cryopreserved PBMCs (2 Years) | Significance |
|---|---|---|---|
| Cell Viability (Post-Thaw/Post-Electroporation) | Baseline | Slight reduction (4.00%–5.67%) [4] | Not statistically significant for T-cell population [4] |
| Early-Stage Proliferation (Day 3 Post-Electroporation) | Baseline | Significantly lower [4] | A key challenge, leading to prolonged culture times |
| Overall Expansion Fold (11-Day Culture) | Baseline | Slight reduction | No significant impact [4] |
| Transfection Efficiency | Baseline | Consistent | No significant difference [4] |
| CD4+/CD8+ T-cell Proportions | Baseline | Consistent | No significant difference [4] |
| T-cell Phenotype (Tn and Tcm) | Baseline | Consistent | No significant difference [4] |
| Cytotoxicity (at E:T Ratio 4:1) | 91.02%–100.00% | 95.46%–98.07% (CAR-2Y) [4] | Comparable tumor-killing capacity [4] |
| Cytokine Secretion Profile | Baseline | Comparable (except transient IFN-γ decrease in CAR-12M) [4] | No systematic changes [4] |
The data demonstrates that while the origin of PBMCs (fresh vs. cryopreserved) has a minimal impact on the final CAR-T product's phenotype and function, the electroporation process itself is the primary driver of the early proliferation delay [4]. Cryopreserved PBMCs show remarkable stability in viability and T-cell composition over long-term storage, making them a viable and logistically superior starting material [4] [51].
Root Cause Analysis: Mechanisms Behind Proliferation Delay
The slower proliferation following non-viral electroporation can be attributed to several interconnected biological mechanisms:
- Cellular Trauma and Membrane Damage: Electroporation employs high electric fields (400–2200 V/cm) to permeabilize the cell membrane [50]. This can create large, unstable pores from which the cell struggles to recover, leading to acute cytotoxicity and the initiation of pro-death signaling pathways that cull the proliferative population [52] [50].
- Persistent Biological Perturbations: Beyond immediate death, sub-lethally injured T cells can experience global changes in gene expression profiles and biological functions. This state diverts cellular resources away from normal growth and division cycles towards damage repair and stress response, effectively inducing a temporary state of proliferative arrest or senescence [50].
- Activation-Induced Cell Death (AICD): Electroporation is typically performed on pre-activated T cells. The combined stress of activation and electrical pulsing can exacerbate AICD, further reducing the pool of cells capable of robust expansion [49].
The diagram below illustrates the cascade of events from electroporation to slowed proliferation.
Optimized Experimental Protocols for Enhanced Proliferation
Addressing the proliferation lag requires a holistic optimization of the entire workflow, from cell source to post-transfection culture. The following protocols are synthesized from recent successful studies.
Protocol 1: PiggyBac Electroporation of Cryopreserved PBMCs
This protocol is adapted from the 2025 comparative study that successfully generated functional CAR-T cells from long-term cryopreserved PBMCs [4].
Step 1: PBMC Thaw and Recovery
- Rapidly thaw cryopreserved PBMCs in a 37°C water bath.
- Dilute the cell suspension drop-wise in pre-warmed complete medium.
- Wash cells to remove residual cryoprotectant (e.g., DMSO) and assess viability.
Step 2: T-Cell Activation
- Isolate T cells using CD4/CD8 magnetic bead enrichment.
- Activate T cells using anti-CD3/CD28 beads or antibodies for 48 hours prior to electroporation. This is a critical window for process optimization.
Step 3: Plasmid Electroporation
- Use the PiggyBac transposon system, consisting of a donor plasmid carrying the CAR transgene and a separate helper plasmid expressing the transposase.
- For electroporation, use a pre-optimized program on a commercial system (e.g., Lonza Nucleofector). The study indicated that optimizing parameters like voltage, pulse length, and buffer composition is key to improving viability.
Step 4: Post-Transfection Recovery and Culture
- Immediately transfer electroporated cells to pre-warmed, cytokine-rich complete medium (e.g., containing IL-2 and IL-7/IL-15).
- This is a critical step. The study emphasized that process optimization, particularly in post-electroporation culture conditions, was more impactful than the fresh/frozen status of PBMCs for enhancing proliferation and toxicity [4].
- Culture cells for 11-14 days, monitoring expansion and phenotype.
Protocol 2: NExT - Nanostraw Electro-Actuated Transfection
The NExT platform represents an advanced physical method designed to minimize cellular damage [50].
Step 1: Platform Preparation
- The system uses a track-etched polyester membrane integrated with millions of hollow, high-aspect-ratio nanostraws.
- The cell suspension is placed on one side of the membrane, while the cargo (e.g., plasmid DNA, mRNA) is in a fluidic reservoir on the other.
Step 2: Nano-Pulsed Electrotransfection
- Apply short, low-energy voltage pulses. The nanostraws act as conduits, locally permeating the cell membrane at the point of contact and enabling electrophoretic delivery of cargo directly into the cytosol.
- This method avoids the widespread, large pore formation associated with bulk electroporation.
Step 3: Post-Transfection Analysis
- This method has demonstrated up to 80% transfection efficiency in primary immune cells with significantly reduced cellular perturbations and higher viability compared to bulk electroporation, creating a healthier starting population for expansion [50].
Table 2: The Scientist's Toolkit: Key Reagents and Materials for Non-Viral CAR-T Manufacturing
| Item Name | Function/Description | Application Note |
|---|---|---|
| PiggyBac Transposon System | A non-viral gene integration system consisting of a donor plasmid and a transposase helper plasmid [4]. | Enables stable genomic integration of large CAR transgenes without viral components. Superior cargo capacity (up to 100 kb) [4]. |
| Cryopreserved PBMCs | Peripheral Blood Mononuclear cells frozen in liquid nitrogen for long-term storage [4] [51]. | Enables batch testing, reduces donor variability, and offers logistical flexibility. T-cell proportions remain stable over long periods [4]. |
| CD3/CD28 Activator Beads | Magnetic beads coated with antibodies to stimulate T-cell activation and proliferation [4]. | Essential pre-stimulation step before genetic engineering. Mimics natural antigen presentation. |
| Lonza Nucleofector System | A widely used electroporation device optimized for hard-to-transfect cells like primary T cells [50]. | Delivers electrical pulses in a specific buffer to facilitate nucleic acid delivery. Requires parameter optimization. |
| Recombinant Human IL-2 | A key T-cell growth factor cytokine [4]. | Added to culture media post-transfection to support T-cell survival, expansion, and function. |
| NExT Platform | A nanostraw-based electrotransfection system for high-throughput, low-trauma gene delivery [50]. | Alternative to bulk electroporation. Can transfect up to 14.4 million cells in a single run with high viability [50]. |
Pathway to Optimization: Strategic Interventions
Overcoming the proliferation bottleneck requires a multi-faceted strategy targeting the key stages of the manufacturing process. The following pathway outlines the main challenges and corresponding solutions.
The choice between fresh and cryopreserved PBMCs is not the primary determinant for the success of non-viral CAR-T manufacturing. As comparative data shows, cryopreserved PBMCs are a logistically advantageous and functionally comparable starting material [4]. The core challenge of slower early-stage proliferation is rooted in the electroporation process itself.
Overcoming this hurdle is achievable through a combination of advanced engineering solutions like NExT technology that minimize cellular damage [50] and rigorous process optimization of pre- and post-transfection protocols, particularly T-cell activation and cytokine-supported recovery [4]. By adopting these strategies, researchers can harness the full potential of non-viral electroporation—its safety, cost-effectiveness, and flexibility—to develop the next generation of accessible and potent CAR-T therapies.
Optimizing Electroporation Parameters for Recovering Cryopreserved T-cells
The transition from lab-scale research to scalable, robust manufacturing is a critical challenge in the field of chimeric antigen receptor (CAR)-T cell therapy [53]. A central question in this process is whether to use fresh or cryopreserved peripheral blood mononuclear cells (PBMCs) as the starting material for T-cell engineering. While fresh PBMCs are often perceived as the gold standard, cryopreserved cells offer significant logistical advantages for centralized manufacturing, including scheduling flexibility, the ability to perform quality control testing on the starting material, and the possibility of collecting cells from patients when they are in a healthier state [8] [7]. The successful use of cryopreserved cells, however, is highly dependent on optimized manufacturing protocols, with electroporation standing out as a key parameter for ensuring high cell viability, transfection efficiency, and ultimately, product functionality [54]. This guide provides a objective comparison of performance data and detailed methodologies for optimizing electroporation in cryopreserved T-cells, framing the discussion within the broader thesis of fresh versus cryopreserved PBMCs for CAR-T manufacturing.
Performance Comparison: Fresh vs. Cryopreserved PBMCs in CAR-T Manufacturing
Extensive research has directly compared the performance of CAR-T cells manufactured from fresh and cryopreserved PBMCs. The collective data indicate that while cryopreservation can introduce some initial changes, the critical functional outputs of the final product remain largely intact.
Table 1: In vitro and Clinical Performance of CAR-T from Fresh vs. Cryopreserved PBMCs
| Performance Metric | Fresh PBMCs | Cryopreserved PBMCs | Significance (p-value) | Study Context |
|---|---|---|---|---|
| Cell Expansion (Fold) | 78.7x ± 37.1 [53] | 158.3x ± 75.3 [53] | Not Reported (N.R.) | Lab-scale, 4 healthy donors [53] |
| Viability (Day 6) | 81.8% ± 7.0 [53] | 94.2% ± 3.7 [53] | N.R. | Lab-scale, 4 healthy donors [53] |
| Transduction Efficiency | Comparable | Comparable | > 0.05 [8] | 118 patient products [8] |
| CD4:CD8 Ratio | Comparable | Comparable | > 0.05 [8] | 118 patient products [8] |
| In vitro Cytotoxicity | High | High, Comparable to Fresh | N.R. [7] | Healthy donors [7] |
| Cytokine Secretion (IFN-γ) | Baseline | Significant decrease in CAR-12M vs. CAR-F [4] | < 0.05 [4] | PiggyBac system [4] |
| 3-Month Complete Response (CR) | 46.2% [5] | 45.5% [5] | > 0.05 [5] | 162 R/R DLBCL patients [5] |
| 1-Year Overall Survival (OS) | 64.1% [5] | 75.4% [5] | > 0.05 [5] | 162 R/R DLBCL patients [5] |
A study on CD19 CAR-T cells for Diffuse Large B-Cell Lymphoma (DLBCL) found no significant differences in clinical outcomes, including overall survival and progression-free survival, between patients treated with products derived from fresh or cryopreserved PBMCs [5]. Similarly, in vitro assays consistently show that CAR-T cells from cryopreserved PBMCs maintain potent anti-tumor cytotoxicity, despite a potential slowing of expansion kinetics during the manufacturing process [7]. One study noted a significant decrease in IFN-γ secretion in CAR-T cells derived from PBMCs cryopreserved for 12 months, though this did not correlate with a loss of cytotoxic function in their model [4].
Electroporation Parameter Optimization: A Comparative Analysis
Electroporation is a critical, non-viral method for introducing genetic material into T-cells. Its success is highly dependent on several interacting parameters, which must be carefully balanced to maximize transfection efficiency while preserving cell health, especially for cryopreserved cells.
Table 2: Impact of Key Electroporation Parameters on T-Cell Engineering Outcomes
| Electroporation Parameter | Impact on Transfection Efficiency | Impact on Cell Viability | Optimized Setting for Cryopreserved T-Cells |
|---|---|---|---|
| Electrical Field Strength | Higher voltage improves plasmid permeation [54] | Inversely correlated with viability; higher voltage decreases viable cells [54] | Must be titrated to find a balance; pulse code CM-138 was identified as optimal in one system [53] |
| Cell Density | Higher cell density can enhance post-electroporation viability for single-activated cells [54] | Critical for recovery; lower densities may improve outcome | ( 1 \times 10^8 ) cells/mL (for 20μL cuvettes, ( 2 \times 10^6 ) cells total) [54] |
| DNA/mRNA Quantity | Higher concentration increases proportion of transfected cells [54] [55] | Higher concentration decreases viability due to DNA toxicity [54] [55] | Requires titration; use minimal amount needed for sufficient expression [55] |
| Cell Activation Status | Stimulation for up to 3 days substantially improves transfection efficiency [54] | Activated cells are more robust to electroporation stress | 1-3 days of activation prior to electroporation is recommended [54] |
| Post-Thaw Resting | Not directly stated | Improves recovery and viability before activation/electroporation | 24 hours of culture before activation is recommended [54] |
The interplay between these parameters is crucial. For instance, increasing the amount of DNA used in electroporation leads to higher transgene expression but at the cost of cell viability, a phenomenon attributed to DNA toxicity [55]. Similarly, while a stronger electrical field improves plasmid uptake, it concurrently increases cell death, necessitating a careful balance [54]. For cryopreserved cells, the post-thaw handling and activation steps become even more critical. Allowing a resting period of at least 24 hours after thawing before activation helps restore cell health and improves subsequent transfection performance [54].
Detailed Experimental Protocols for Electroporation
Protocol for T-Cell Activation & Expansion from Cryopreserved PBMCs
This protocol is adapted from methods used in comparative studies of fresh and cryopreserved starting materials [53] [54].
- Thawing and Washing: Rapidly thaw cryopreserved PBMCs in a 37°C water bath. Immediately transfer the cells to a pre-warmed culture medium (e.g., OpTmizer CTS or X-VIVO) supplemented with human serum or fetal bovine serum (FBS). Centrifuge to remove the cryoprotectant (e.g., DMSO) and resuspend the cell pellet in fresh, pre-warmed complete medium.
- Resting: Culture the washed PBMCs overnight (approximately 24 hours) at 37°C in a humidified 5% CO2 incubator. This resting period is critical for allowing cellular recovery from cryopreservation stress [54].
- T-Cell Isolation (Optional): Isolate T-cells from the PBMCs using negative selection magnetic bead kits (e.g., EasySep Human T Cell Isolation Kit) according to the manufacturer's instructions [7].
- Activation: Activate the T-cells using anti-CD3/anti-CD28 activator reagents. The format (soluble antibodies vs. immobilized on beads or a nanomatrix) can impact initial cell surface marker profiles and expansion [53]. A common method is using Dynabeads CD3/CD28 at a bead-to-cell ratio of 1:3 [54].
- Expansion: Culture the activated T-cells in complete medium supplemented with recombinant human IL-2 (e.g., 100-300 IU/mL). Maintain cell concentrations between ( 0.5 - 2.0 \times 10^6 ) cells/mL and split as necessary. The optimal transfection window is typically between days 6-10 of expansion [53].
Optimized DNA/mRNA Electroporation Protocol
This protocol synthesizes optimized steps from multiple studies for transfecting activated T-cells [53] [54] [55].
- Preparation: On the day of electroporation, harvest activated T-cells and remove activation beads using a magnet. Perform a cell count and viability assessment.
- DNA/mRNA Preparation: Ensure nucleic acids are of high quality. For DNA, use endotoxin-free kits and resuspend in water (not TE buffer, as EDTA interferes with electroporation). The OD260/280 ratio should be >1.8, with most plasmid in a supercoiled conformation [55].
- Electroporation Setup: Wash cells and resuspend them in an appropriate electroporation buffer at the optimized density (e.g., ( 1 \times 10^8 ) cells/mL). Combine the cell suspension with the predetermined optimal amount of DNA or mRNA in an electroporation cuvette. Keep the cuvette on ice.
- Electroporation: Transfer the cuvette to the electroporator and apply the optimized electrical parameters. For example, one study identified "pulse code CM-138" on a specific system as optimal for mRNA, yielding high transgene expression and preserved viability [53].
- Post-Electroporation Recovery: Immediately after pulsing, transfer the cells from the cuvette into pre-warmed complete medium. Do not rinse the cuvette, as this can damage cells. Incubate the cells at 37°C for at least 20 minutes to allow membrane resealing before further handling or plating [55].
- Cryopreservation of Electroporated Cells (Optional): For creating "Assay Ready" cell banks, cells can be cryopreserved after the recovery period. Cells are typically frozen in a solution containing 10% DMSO and 20-90% serum, or a commercial cryopreservation medium, using a controlled-rate freezer before long-term storage in liquid nitrogen [56] [55].
Logical Workflow for Process Optimization
The following diagram synthesizes the key decision points and procedural steps for optimizing electroporation of cryopreserved T-cells, from material preparation to final analysis.
The Scientist's Toolkit: Essential Reagents & Materials
Successful electroporation and culture of cryopreserved T-cells relies on a suite of specialized reagents and instruments.
Table 3: Key Research Reagent Solutions for T-Cell Electroporation
| Item Category | Specific Examples | Function & Application Notes |
|---|---|---|
| Cell Culture Media | ImmunoCult-XF T Cell Expansion Medium, TheraPEAK T-VIVO, OpTmizer CTS, AIM-V [53] [54] [8] | Supports T-cell activation and expansion; formulation can significantly impact expansion rates and viability [53]. |
| Activation Reagents | Dynabeads CD3/CD28 CTS, Soluble anti-CD2/anti-CD3/anti-CD28 [53] [54] | Provides the critical signal 1 (CD3) and signal 2 (CD28) for T-cell activation. Format (soluble vs. immobilized) affects CD3 internalization and phenotype [53]. |
| Electroporation Systems | Celetrix system, MaxCyte ExPERT platform [54] [56] | Instruments that deliver controlled electrical pulses. Performance is system-specific, requiring optimization of proprietary pulse codes or programs [53] [56]. |
| Electroporation Buffers | System-specific proprietary buffers | Maintains ionic balance and conductivity during electrical pulse to facilitate nucleic acid entry with minimal cell damage. |
| Genetic Cargo | In vitro transcribed (IVT) mRNA, Plasmid DNA (e.g., pmaxGFP, pcDNA3.1-CAR) [53] [54] | mRNA enables transient, high-level expression with lower genotoxic risk. DNA is used for non-viral transposon systems (e.g., PiggyBac) for stable integration [4] [54]. |
| Cryopreservation Media | CryoSure-DEX40, Synth-a-Freeze CTS, Human serum with 5-10% DMSO [5] [7] | Protects cells from ice crystal formation and osmotic shock during freezing and thawing, ensuring high post-thaw viability. |
The body of evidence strongly supports the viability of cryopreserved PBMCs as a starting material for CAR-T cell manufacturing. While initial cell expansion may be slower and subtle phenotypic differences may exist, the critical functional outcomes—including transduction efficiency, in vitro cytotoxicity, and most importantly, clinical efficacy in patients—are not significantly compromised by the use of frozen cells [5] [8] [7]. The key to unlocking the performance of cryopreserved T-cells lies in a rigorous, systematic optimization of the electroporation process. By carefully balancing electrical parameters, cell health, and nucleic acid quality, researchers and manufacturers can leverage the considerable logistical advantages of cryopreservation without sacrificing product quality. This approach makes advanced cell therapies more accessible and adaptable to a global manufacturing landscape.
Minimizing Cell Exhaustion and Maintaining Stemness During Prolonged Culture
The transition to using cryopreserved peripheral blood mononuclear cells (PBMCs) as starting material for Chimeric Antigen Receptor T-cell (CAR-T) manufacturing represents a significant paradigm shift in cellular immunotherapy. This comprehensive analysis compares the performance of CAR-T cells generated from cryopreserved versus fresh PBMCs, with a specific focus on critical quality attributes including T-cell exhaustion, differentiation status, and stemness preservation during extended in vitro culture. Current evidence demonstrates that through optimized protocols, cryopreserved PBMCs can yield CAR-T products with comparable expansion potential, phenotypic characteristics, and cytotoxic functionality to their fresh counterparts, while effectively maintaining favorable differentiation profiles essential for long-term persistence and efficacy. This review synthesizes experimental data, methodological approaches, and technological innovations that enable the use of cryopreserved cells without compromising product quality, thereby addressing key logistical challenges in distributed CAR-T manufacturing.
Autologous CAR-T cell therapy has revolutionized cancer treatment, particularly for hematological malignancies, yet faces significant logistical challenges that impact patient access and treatment efficacy. The current reliance on fresh patient cells creates manufacturing bottlenecks, with vein-to-vein times typically ranging from 3-5 weeks [57]. This delay poses a critical barrier for patients with aggressive diseases, with studies indicating that nearly 30% of patients prescribed CAR-T therapy never undergo leukapheresis, and 20% of those who do undergo apheresis never receive infusion due to disease progression or clinical deterioration [57].
Cryopreserved PBMCs offer a potential solution to these challenges by decoupling cell collection from manufacturing. This approach enables the creation of cell banks from healthy donors or patients during optimal health status, potentially mitigating issues associated with T-cell deterioration following extensive preconditioning therapies [4]. Furthermore, evidence suggests that T-cells isolated from healthy individuals are less differentiated and better retain stem-like characteristics during culture, which may enhance the therapeutic efficacy of the resulting CAR-T products [4].
While lentiviral processes for generating CAR-T from cryopreserved PBMCs have been reported, successful non-viral approaches using transposon systems like PiggyBac have been less documented [4] [58]. This review systematically evaluates the comparative performance of CAR-T cells manufactured from cryopreserved versus fresh PBMCs, with particular emphasis on strategies to minimize exhaustion and maintain stemness during prolonged culture periods essential for commercial manufacturing.
Comparative Performance Analysis: Cryopreserved vs. Fresh PBMCs
Impact of Cryopreservation Duration on PBMC Viability and Phenotype
Long-term cryopreservation maintains PBMC viability and critical T-cell subsets necessary for CAR-T manufacturing. Studies evaluating PBMCs cryopreserved for periods ranging from 3 months to 3.5 years demonstrate that although a statistically significant decrease in viability occurs compared to fresh cells, the actual reduction is only 4.00% to 5.67%, with average viability remaining at approximately 90.95% even after 3.5 years of storage [4]. More importantly, the proportion of T-cells remains relatively stable post-cryopreservation, while natural killer (NK) cells and B cells show decreased proportions due to their heightened sensitivity to hypothermic conditions [4].
Table 1: Viability and Phenotypic Stability of PBMCs After Long-Term Cryopreservation
| Cryopreservation Duration | Average Viability (%) | T-cell Population Stability | Naïve T-cell (Tn) Maintenance | Central Memory T-cell (Tcm) Maintenance |
|---|---|---|---|---|
| Fresh PBMCs | 95.67 (reference) | Stable (reference) | Stable (reference) | Stable (reference) |
| 3 months | 91.67 | Comparable to fresh | No significant change | No significant change |
| 6 months | 90.67 | Comparable to fresh | No significant change | No significant change |
| 12 months | 91.00 | Comparable to fresh | No significant change | No significant change |
| 2 years | 90.00 | Comparable to fresh | No significant change | No significant change |
| 3.5 years | 90.95 | Not reported | Not reported | Not reported |
Crucially, the T-cell differentiation state—particularly the proportions of naïve T-cells (Tn) and central memory T-cells (Tcm)—remains unchanged following cryopreservation compared to fresh samples [4]. These cell populations are essential for CAR-T efficacy as they enhance activation, persistence, and effector function, ultimately improving therapeutic outcomes [4].
CAR-T Product Characteristics from Cryopreserved vs. Fresh PBMCs
Comprehensive comparative studies reveal that CAR-T cells generated from cryopreserved PBMCs exhibit comparable critical quality attributes to those derived from fresh cells. Research utilizing the PiggyBac transposon system for mesothelin-targeting CAR (mesoCAR-T) demonstrated no significant differences in expansion potential, cell phenotype, or transfection efficiency between products derived from fresh versus cryopreserved PBMCs, even after 2 years of frozen storage [4].
Table 2: Functional Comparison of mesoCAR-T Cells from Fresh vs. Cryopreserved PBMCs
| Performance Metric | Fresh PBMCs-derived CAR-T | Cryopreserved PBMCs-derived CAR-T (2 years) | Statistical Significance |
|---|---|---|---|
| Cell Viability | Comparable baseline | Comparable baseline | Not significant |
| Proliferation Capacity | Reference expansion | Slight reduction, not impactful | Not significant |
| CD3+ Purity | Stable during culture | Stable during culture | Not significant |
| CD4+/CD8+ Ratio | Stable during culture | Stable during culture | Not significant |
| Transfection Efficiency | Comparable | Comparable | Not significant |
| Tn/Tcm Maintenance | Gradual decrease with culture | Similar pattern to fresh | Not significant |
| Exhaustion Markers | Baseline expression | Comparable expression | Not significant |
| Cytotoxicity (4:1 E:T) | 91.02%–100.00% | 95.46%–98.07% | Not significant |
| Cytotoxicity (2:1 E:T) | Comparable baseline | Comparable baseline | Not significant |
| Cytokine Secretion Profile | Reference pattern | Comparable, except IFN-γ (decreased in CAR-12M) | Significant for IFN-γ only |
Functional assessments confirm that mesoCAR-T cells from cryopreserved PBMCs maintain potent antitumor activity. Cytotoxicity against human ovarian cancer cell line (SKOV-3) cells was comparable between groups, with cryopreserved-derived products exhibiting 95.46%–98.07% specific lysis at 4:1 effector-to-target ratios versus 91.02%–100.00% for fresh-derived products [4]. Cytokine secretion profiles were largely similar, though a significant decrease in IFN-γ was observed in cells derived from 12-month cryopreserved PBMCs without compromising cytotoxic function [4].
Methodological Framework: Experimental Protocols for Comparative Analysis
Standardized Cryopreservation Protocol for PBMCs
Optimal cryopreservation methodology is fundamental to preserving T-cell fitness and functionality. The following protocol has been validated for maintaining PBMC viability and function across extended storage periods:
Materials Required:
- Cryopreservation medium: CryoStor CS10 (serum-free) or 90% FBS + 10% DMSO
- Cryogenic vials
- Controlled-rate freezer or isopropanol freezing container (e.g., CoolCell)
- Pre-chilled centrifuge
Procedure:
- Centrifuge PBMCs at 300 × g for 10 minutes to pellet cells [19].
- Carefully aspirate supernatant without disturbing cell pellet.
- Resuspend cell pellet in cold (2-8°C) cryopreservation medium at recommended concentration of 0.5-10 × 10⁶ cells/mL [19].
- For DMSO/FBS medium: Pre-mix cells with FBS first, then add DMSO to final 10% concentration gradually to minimize osmotic shock.
- Aliquot 1 mL cell suspension into pre-chilled cryovials.
- Initiate controlled-rate freezing at approximately -1°C/minute using specialized equipment or isopropanol containers placed at -80°C overnight [19].
- Transfer cryovials to vapor-phase liquid nitrogen for long-term storage below -135°C [19].
Critical Considerations:
- Limit time between cryoprotectant addition and freezing initiation to ≤120 minutes [3].
- Avoid storage at -80°C for extended periods as this compromises viability.
- Use serum-free, defined cryomedium like CryoStor CS10 for clinical applications to minimize batch variability and pathogen transmission risks [11].
CAR-T Generation via PiggyBac Transposon System
The non-viral PiggyBac transposon system offers advantages for CAR-T manufacturing, including reduced costs, increased cargo capacity, and lower immunogenicity compared to viral systems [4]. The following methodology has been successfully applied to both fresh and cryopreserved PBMCs:
Workflow:
- Thawing and Recovery: Rapidly thaw cryopreserved PBMCs in a 37°C water bath, then transfer to pre-warmed culture medium containing DNase (10 µg/mL) to prevent cell clumping [11].
- T-cell Enrichment: Isolate CD4+/CD8+ T-cells using magnetic bead separation.
- T-cell Activation: Stimulate T-cells with anti-CD3/CD28 antibodies for 48 hours.
- Electroporation: Introduce PiggyBac transposon system containing CAR construct via electroporation.
- Ex Vivo Expansion: Culture transfected cells for 11 days with appropriate cytokine support.
- Quality Assessment: Monitor cell expansion, phenotype, and functionality throughout culture.
Process Optimization: Studies indicate that post-electroporation viability dip on day 3 represents a critical process challenge. Targeted optimization of activation conditions, cytokine supplementation, and culture parameters can enhance recovery and expansion while maintaining favorable differentiation profiles [4].
Diagram 1: Experimental workflow for comparative CAR-T manufacturing from fresh versus cryopreserved PBMCs using the PiggyBac transposon system.
Assessment Methodologies for Exhaustion and Stemness
Comprehensive evaluation of T-cell exhaustion and differentiation status employs multiple complementary techniques:
Flow Cytometric Analysis:
- Differentiation Status: CD45RO and CCR7 staining to identify Tn (CD45RO-CCR7+), Tcm (CD45RO+CCR7+), Tem (CD45RO+CCR7-), and Temra (CD45RO-CCR7-) populations [4].
- Exhaustion Markers: PD-1, TIM-3, LAG-3, TIGIT expression profiling [4] [59].
- Stemness Markers: TCF1 (TCF7) detection to identify progenitor-like subsets [59].
Functional Assays:
- Cytotoxicity: Real-time cellular analysis (RTCA) against target tumor cells (e.g., SKOV-3) at multiple effector-to-target ratios [4].
- Cytokine Secretion: Multiplex analysis of IFN-γ, IL-2, TNF-α, IL-6, IL-10, and other relevant cytokines [4].
- Proliferation Capacity: CFSE or similar dye dilution assays to track expansion potential.
Advanced Molecular Techniques:
- Proteomic Analysis: Mass spectrometry to identify proteotoxic stress response signatures associated with exhaustion [60].
- Epigenetic Profiling: ATAC-seq and histone modification analysis to examine chromatin accessibility and epigenetic locking [59].
Mechanisms of T-cell Exhaustion and Stemness Regulation
Molecular Drivers of T-cell Exhaustion
T-cell exhaustion represents a hypofunctional state characterized by reduced effector function and increased inhibitory receptor expression that arises from persistent antigen exposure and hostile microenvironmental signals [60]. Recent research has identified a distinct proteotoxic stress response (Tex-PSR) as a hallmark and mechanistic driver of exhaustion. Contrary to canonical stress responses that reduce protein synthesis, Tex-PSR involves increased global translation activity, upregulation of specialized chaperone proteins, accumulation of protein aggregates, and enhanced autophagy-dominant protein catabolism [60].
The exhaustion program generates heterogeneous T-cell populations. Progenitor exhausted T (Tprog) cells retain stemness and self-renewal capacity and respond to immune checkpoint blockade therapies, while terminal exhausted T (Ttex) cells accumulate over time and respond poorly to interventions [60] [59]. The transition between these states is regulated by epigenetic mechanisms that create stable repressive chromatin landscapes, effectively "locking in" the exhausted phenotype through DNA methylation, histone modifications, and non-coding RNA networks [59].
Diagram 2: Molecular pathways driving T-cell exhaustion and potential intervention points for culture optimization.
Strategic Approaches to Minimize Exhaustion in Prolonged Culture
Culture duration significantly impacts T-cell differentiation and exhaustion states. Studies consistently show that shortened culture times better preserve stem-like memory phenotypes, which correlate with enhanced persistence and antitumor efficacy in vivo [4]. Several strategic approaches can mitigate exhaustion during manufacturing:
Process Parameter Optimization:
- Culture Duration: Limit ex vivo expansion periods where feasible without compromising yield requirements.
- Activation Conditions: Titrate stimulation intensity to achieve sufficient activation while minimizing differentiation pressure.
- Cytokine Supplementation: Utilize cytokines such as IL-7 and IL-15 that support memory phenotypes rather than promoting terminal differentiation.
Metabolic and Epigenetic Modulation:
- Metabolic Programming: Maintain oxidative rather than purely glycolytic metabolism to support memory cell formation.
- Epigenetic Interventions: Target exhaustion-associated epigenetic regulators such as DNMTs, HDACs, EZH2, or BET family proteins to prevent or reverse epigenetic locking [59].
Alternative Manufacturing Platforms:
- Rapid Manufacturing: Emerging rapid CAR-T production platforms (2-3 days versus conventional 7-11 days) demonstrate reduced exhaustion markers and enhanced stemness preservation [57].
Research Reagent Solutions for Exhaustion and Stemness Research
Table 3: Essential Research Tools for T-cell Exhaustion and Stemness Investigations
| Reagent Category | Specific Products | Research Application | Functional Role |
|---|---|---|---|
| Cryopreservation Media | CryoStor CS10, NutriFreez D10 | Long-term PBMC storage | Serum-free alternatives maintaining viability and function [11] |
| Cell Separation | CD4+/CD8+ magnetic beads | T-cell enrichment | Isolation of target populations from PBMCs or leukapheresis [4] |
| Activation Reagents | Anti-CD3/CD28 antibodies | T-cell activation | Mimic antigen presentation and costimulation [4] |
| Genetic Modification | PiggyBac transposon system | Non-viral CAR insertion | Cost-effective gene delivery with large cargo capacity [4] |
| Cytokine Cocktails | IL-2, IL-7, IL-15 | Culture supplementation | Support expansion and influence differentiation fate [4] |
| Exhaustion Markers | Anti-PD-1, TIM-3, LAG-3 antibodies | Phenotypic characterization | Detect and quantify exhaustion states [4] [59] |
| Differentiation Markers | Anti-CD45RO, CCR7 antibodies | Memory subset identification | Distinguish Tn, Tcm, Tem, and Temra populations [4] |
| Stemness Markers | Anti-TCF1 (TCF7) antibodies | Progenitor population identification | Detect self-renewing T-cell subsets [59] |
The comprehensive analysis of current evidence demonstrates that cryopreserved PBMCs serve as a viable alternative to fresh cells for CAR-T manufacturing, with comparable performance across critical quality attributes including expansion potential, phenotype, and antitumor functionality. Through optimized protocols addressing cryopreservation methods, manufacturing processes, and culture conditions, it is feasible to generate CAR-T products from cryopreserved starting materials while minimizing exhaustion and preserving beneficial stem-like memory populations.
The strategic implementation of cryopreserved PBMCs addresses significant logistical challenges in CAR-T production, potentially expanding patient access through enhanced supply chain flexibility and enabling the use of cells collected during optimal health status. Future directions should focus on further standardization of cryopreservation protocols, validation across diverse manufacturing platforms, and clinical correlation of these findings to establish cryopreserved starting materials as a new standard in distributed CAR-T manufacturing.
Managing Donor Variability and Pre-Cryopreservation Cell Health
The transition to using cryopreserved peripheral blood mononuclear cells (PBMCs) as starting material for Chimeric Antigen Receptor T-cell (CAR-T) manufacturing represents a significant shift in immunotherapy production paradigms. This evolution addresses critical challenges in donor variability and pre-cryopreservation cell health that directly impact product consistency and therapeutic outcomes. For researchers and drug development professionals, understanding the quantitative and functional implications of this transition is essential for optimizing CAR-T manufacturing processes.
Cryopreserved PBMCs offer practical advantages over fresh cells by decoupling cell collection from manufacturing, enabling standardized testing, and facilitating the use of healthy donor cells collected at optimal health stages [4]. However, the process introduces unique considerations for managing donor-specific characteristics and preserving cellular integrity through freeze-thaw cycles. This guide provides a comparative analysis of critical quality attributes between fresh and cryopreserved PBMCs, with experimental data to inform protocol development and raw material selection for CAR-T manufacturing.
Comparative Performance Analysis: Fresh vs. Cryopreserved PBMCs
Viability and Stability Metrics
Table 1: Viability and Composition Stability of Cryopreserved PBMCs
| Parameter | Fresh PBMCs | Cryopreserved (3-24 months) | Significance | Source |
|---|---|---|---|---|
| Viability | ~99.0% | 90.9%-97.0% (post-thaw) | Minimal decline (4.00%-5.67%) post-thaw | [4] [3] |
| T-cell Proportion | Baseline | Remains stable | No significant loss of CD3+ T cells | [4] |
| Lymphocyte Percentage | 52.20% (PBMCs) | 66.59% (Cryo-LEUK) | Higher in cryopreserved leukapheresis | [3] |
| Naïve/Memory T-cells | Baseline proportions maintained | Tn and Tcm populations preserved | Critical for CAR-T persistence | [4] |
| Long-term Stability | N/A | Viability stable up to 3.5 years | Viability: ~90.95% at 3.5 years | [4] |
The data demonstrates that cryopreservation maintains sufficient viability and preserves key T-cell subpopulations necessary for effective CAR-T manufacturing. The higher lymphocyte percentage in cryopreserved leukapheresis products suggests potential advantages for T-cell based therapies [3].
Functional Performance in CAR-T Manufacturing
Table 2: CAR-T Functional Outcomes from Fresh vs. Cryopreserved PBMCs
| Functional Attribute | Fresh PBMCs | Cryopreserved PBMCs | Significance | Source |
|---|---|---|---|---|
| Expansion Potential | Baseline | Comparable | No significant impact on proliferation | [4] |
| Transfection Efficiency | Baseline | Comparable (PiggyBac system) | Successful non-viral transfection demonstrated | [4] |
| Cytotoxicity | 91.02%-100% (at E:T 4:1) | 95.46%-98.07% (at E:T 4:1) | Comparable tumor cell killing | [4] |
| Cytokine Secretion | Baseline pattern | Comparable (except IFN-γ in CAR-12M) | No systematic changes in cytokine profile | [4] |
| Exhaustion Markers | Baseline | Comparable | No significant differences in persistence markers | [4] |
| Platform Compatibility | Compatible with multiple platforms | Validated for viral, non-viral, and Fast CAR-T platforms | No compromise in consistency across systems | [3] |
The functional comparability between CAR-T products generated from fresh and cryopreserved PBMCs supports the feasibility of cryopreserved starting materials, even with the non-viral PiggyBac electroporation system [4].
Experimental Protocols for Comparative Analysis
Cryopreservation and Thawing Methodology
Optimized Cryopreservation Protocol:
- Cell Preparation: Isolate PBMCs using density gradient centrifugation (Lymphoprep) at room temperature to ensure proper blood separation [61].
- Cryoprotectant Formulation: Use clinical-grade cryoprotectant containing 10% DMSO in serum-free media such as CryoStor CS10 or NutriFreez D10 [31].
- Freezing Parameters: Resuspend cells at 5×10⁷–8×10⁷ cells/mL in cryoprotectant. Use controlled-rate freezing at approximately -1°C/minute to -90°C, then transfer to vapor-phase liquid nitrogen for storage [61] [3].
- Time Sensitivity: Limit time from cryoprotectant addition to freezing initiation to ≤120 minutes to maintain cell viability [3].
Thawing and Recovery Protocol:
- Rapid Thawing: Thaw cells in 37°C water bath until small ice crystal remains [22].
- Gradual Dilution: Transfer cell suspension to pre-warmed RP10 medium (RPMI1640 with 10% FBS, 10mM HEPES, and gentamycin) [22].
- Centrifugation: Wash cells twice at 500×g for 5 minutes at room temperature to remove cryoprotectant [22].
- Viability Assessment: Determine viability using trypan blue exclusion or propidium iodide staining before proceeding to CAR-T manufacturing [22].
CAR-T Generation and Functional Assays
PiggyBac Transposon System Protocol:
- T-cell Activation: Isolate T-cells using CD4/CD8 magnetic bead enrichment 48 hours post-thawing [4].
- Electroporation: Introduce MSLN CAR vector via PiggyBac electroporation system [4].
- Ex Vivo Culture: Culture cells for 11 days to generate mesoCAR-T cells with regular monitoring of expansion and phenotype [4].
Functional Assessment Methods:
- Cytotoxicity Assay: Use real-time cellular analysis (RTCA) against human ovarian cancer cell line (SKOV-3) at effector-to-target ratios of 4:1 and 2:1 [4].
- Phenotypic Characterization: Employ multicolor flow cytometry to assess T-cell differentiation markers (CD45RO, CCR7) and exhaustion markers [4].
- Cytokine Profiling: Measure secretion of IFN-γ, IL-6, IL-10, IL-5, IL-4, IL-13, IL-2, and TNF-α using multiplex assays [4].
- Single-Cell RNA Sequencing: Apply scRNA-seq to evaluate transcriptomic profiles and cellular heterogeneity after cryopreservation [22].
Figure 1: CAR-T Manufacturing Workflow from Cryopreserved PBMCs. The process highlights critical checkpoints for managing donor variability and cell health assessment.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Cryopreservation and CAR-T Manufacturing
| Reagent Category | Specific Products | Function & Importance | Optimization Notes |
|---|---|---|---|
| Cryopreservation Media | CryoStor CS10, NutriFreez D10, Bambanker D10 | Cryoprotection with 10% DMSO; maintains viability and functionality | Serum-free alternatives eliminate FBS variability [31] |
| Separation Media | Lymphoprep, Ficoll, Histopaque | PBMC isolation via density gradient centrifugation | Room temperature operation critical for proper separation [61] |
| Cell Activation | CD3/CD28 magnetic beads | T-cell activation pre-genetic modification | 48-hour activation post-thaw optimal for cryopreserved PBMCs [4] |
| Genetic Modification | PiggyBac transposon system, Lentiviral vectors | CAR gene delivery | PiggyBac reduces cost with comparable efficiency [4] |
| Culture Media | RPMI-1640 with supplements | Cell expansion and maintenance | HEPES buffer and gentamycin improve recovery [22] |
| Viability Assessment | Trypan blue, Propidium iodide, Live/Dead fixable stains | Pre- and post-cryopreservation quality control | Multiple methods validate recovery [22] |
Technical Considerations for Process Optimization
Managing Donor Variability
Donor variability remains a significant challenge in CAR-T manufacturing, influencing cell recovery and performance. Several strategies can mitigate this variability:
- Donor Screening Programs: Implement verified donor programs with recallable donor pools to ensure consistency in age, health, and collection conditions [61].
- Pre-collection Health Assessment: Establish criteria for donor eligibility that minimizes underlying health conditions affecting T-cell fitness [4].
- Standardized Collection Protocols: Use consistent apheresis parameters and collection containers with appropriate anticoagulants to reduce technical variability [61].
- Cell Composition Monitoring: Regularly assess PBMC composition across donors, noting that cryopreserved leukapheresis products show higher lymphocyte percentages (66.59%) versus cryopreserved PBMCs (52.20%) [3].
Preserving Pre-Cryopreservation Cell Health
Maintaining cell health before cryopreservation requires attention to multiple factors in the collection and processing workflow:
- Time Sensitivity: Process and cryopreserve cells within 24 hours of collection for optimal recovery [61].
- Temperature Control: Maintain room temperature (15-25°C) during transport and processing to prevent granulocyte contamination [61].
- Cryoprotectant Exposure: Limit DMSO exposure time to <120 minutes before freezing to minimize cytotoxicity [3].
- Controlled-Rate Freezing: Implement consistent freezing rates of approximately -1°C/minute to prevent intracellular ice crystal formation [61].
Figure 2: Factors Influencing Donor Variability and Cell Health. The diagram illustrates the relationship between donor characteristics, processing protocols, and final CAR-T product performance.
The comprehensive comparison between fresh and cryopreserved PBMCs for CAR-T manufacturing reveals minimal significant differences in critical quality attributes when optimized protocols are implemented. Cryopreserved PBMCs maintain viability, expansion potential, phenotype characteristics, and cytotoxic functionality comparable to fresh cells, supporting their adoption as a standardized starting material.
The key to managing donor variability and pre-cryopreservation cell health lies in standardized protocols from collection through freezing and thawing. The use of serum-free cryopreservation media, controlled-rate freezing, and rapid processing workflows ensures consistent recovery and performance. Furthermore, the compatibility of cryopreserved PBMCs with multiple CAR-T manufacturing platforms, including non-viral PiggyBac systems, provides flexibility in process design.
For researchers and therapy developers, establishing rigorous donor screening programs, implementing validated cryopreservation protocols, and conducting comprehensive functional assessments throughout development will ensure the consistent manufacturing of high-quality CAR-T products from cryopreserved PBMCs.
Head-to-Head Comparisons: Functional and Clinical Outcomes of CAR-T Products
The choice between using fresh or cryopreserved peripheral blood mononuclear cells (PBMCs) is a critical consideration in the manufacturing of chimeric antigen receptor T-cell (CAR-T) therapies. This guide provides an objective comparison of these starting materials by synthesizing experimental data on cytotoxicity, cell expansion, and cytokine secretion profiles. The consistent performance of cryopreserved PBMCs across these key functional parameters supports their use as a reliable and flexible alternative, potentially revolutionizing CAR-T production models by enabling the use of healthier donor cells and offering greater logistical flexibility [4] [58] [5].
Comparative Performance Data: Fresh vs. Cryopreserved PBMCs
Cell Expansion and Phenotype
Table 1: Comparison of Expansion Potential and Phenotypic Markers
| Parameter | Fresh PBMCs | Cryopreserved PBMCs | Significance |
|---|---|---|---|
| Expansion Fold | Baseline | Comparable [4] [58] | No significant decline even after 2 years of cryopreservation [4] [58]. |
| Viability Post-Thaw | - | ~90-95% [4] | Minimal impact from long-term cryopreservation. |
| T Cell Proportion | Stable baseline | Remains stable post-cryopreservation [4] | Key for CAR-T manufacturing. |
| CD4+/CD8+ Ratio | Baseline | Unaffected [4] | Maintains balance in final CAR-T product. |
| Tn (Naïve) & Tcm (Central Memory) Cells | Baseline | No significant changes [4] | Crucial for long-term CAR-T persistence and efficacy. |
Cytotoxic Function and Cytokine Secretion
Table 2: Comparison of Functional Outputs
| Parameter | Fresh PBMCs | Cryopreserved PBMCs | Significance |
|---|---|---|---|
| Cytotoxicity (at E:T 2:1) | Baseline (91-100%) | Comparable (95-98%) [4] [58] | No statistically significant difference. |
| In Vivo Persistence (Median Duration) | 21 days | 21 days [5] | Identical performance in patients. |
| IFN-γ Secretion | Baseline | Slight decrease in one study [4] | No associated impact on cytotoxic function [4]. |
| Other Cytokines (IL-2, TNF-α, etc.) | Baseline | No systematic changes [4] | Comparable secretion profiles. |
| Clinical Response (1-year OS/PFS) | Comparable (OS: 64.1%, PFS: 44.5%) | Comparable (OS: 75.4%, PFS: 52.1%) [5] | No significant difference in DLBCL patients. |
Detailed Experimental Protocols for Functional Assays
CAR-T Cell Generation from PBMCs
The following protocol, adapted from Scientific Reports, allows for direct comparison between fresh and cryopreserved PBMCs [4].
Workflow Overview: CAR-T Cell Manufacturing
Key Steps:
- PBMC Isolation: Isolate PBMCs from whole blood using Ficoll-Hypaque density gradient centrifugation [4] [62].
- Cryopreservation: Freeze PBMCs using a controlled-rate freezer and store in the vapor phase of liquid nitrogen. Cryopreserved PBMCs can be stored for several years while maintaining high viability and function [4] [5].
- Thawing and T Cell Activation: Thaw cryopreserved PBMCs rapidly and activate them using anti-CD3/CD28 magnetic beads for 48 hours [4].
- Genetic Modification: Introduce the CAR transgene using the PiggyBac transposon system via electroporation. This non-viral method is cost-effective and has a large cargo capacity [4] [58].
- Ex Vivo Expansion: Culture the modified T cells for approximately 11 days in media supplemented with interleukin-2 (IL-2) to achieve the desired expansion fold [4].
Cytotoxicity Assay (Real-Time Cellular Analysis)
This assay quantitatively measures the ability of generated CAR-T cells to kill target cancer cells.
Workflow Overview: Cytotoxicity Assay
Key Steps:
- Seed Target Cells: Plate adherent target cells (e.g., SKOV-3 ovarian cancer cells) in specialized microelectronic sensor (E-plate) plates [4].
- Co-culture: Add the effector CAR-T cells at various effector-to-target (E:T) ratios (e.g., 4:1 and 2:1) [4].
- Real-Time Monitoring: Place the plate in the RTCA instrument. The system continuously measures electrical impedance across the well, which correlates directly with cell number and viability [4].
- Data Analysis: Cytotoxicity is calculated based on the reduction in impedance (indicating target cell death) in experimental wells compared to control wells containing only target cells. The results are expressed as a percentage of target cell lysis [4].
Cytokine Secretion Profiling
This protocol assesses the functional potency and immune activation of CAR-T cells by measuring secreted cytokines.
Key Steps:
- Sample Collection: Collect supernatant from CAR-T and target cell co-cultures after a defined incubation period (e.g., 24 hours) [4].
- Multiplex Immunoassay: Use a multiplex bead-based array (e.g., Luminex) or ELISA to simultaneously quantify the concentrations of multiple cytokines, including:
Enhancing Cytotoxicity via Cytokine Conditioning
Research on Vγ9Vδ2 T cells demonstrates that the cytokine combination used during expansion directly influences cytotoxic potential. Expansion with a combination of IL-2 and IL-15 yields cells with enhanced cytotoxicity compared to IL-2 alone. This is correlated with increased expression and release of cytotoxic molecules like perforin, granzyme B, and granulysin, and higher levels of the transcription factor T-bet [63]. This principle can be applied to CAR-T manufacturing to improve the potency of the final product.
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for CAR-T Functional Assays
| Reagent / Solution | Function / Purpose | Examples / Notes |
|---|---|---|
| Cell Culture Media | Supports cell growth and viability. | X-Vivo 15 [63]; RPMI-1640 or DMEM, often supplemented with human serum or platelet lysate for GMP compliance [62]. |
| Human Serum (HS) / Platelet Lysate (hPL) | Serum supplement for cell culture; provides essential growth factors. | Preferable to FBS for clinical-grade manufacturing. hPL may support higher expansion rates [62]. |
| Recombinant Human Cytokines | Drives T-cell expansion, survival, and can enhance function. | IL-2: Standard expansion [63] [62]. IL-15: Can be combined with IL-2 to enhance cytotoxicity and T-bet expression [63]. IFN-γ: Used in CIK cell generation protocols [62]. |
| Activation Agents | Stimulates T-cell activation prior to genetic modification. | Anti-CD3 antibody [62]; Zoledronic Acid (ZOL) for Vγ9Vδ2 T cell expansion [63]. |
| Transfection System | Introduces genetic material (CAR) into T cells. | PiggyBac Transposon System: Non-viral, cost-effective alternative with high cargo capacity [4] [58]. |
| Cryopreservation Medium | Protects cells during freezing and long-term storage. | Typically contains a cryoprotectant like DMSO (e.g., 10%) and a protein source [63] [4]. |
The comprehensive experimental data from cytotoxicity, expansion, and cytokine secretion assays demonstrate that cryopreserved PBMCs are a functionally equivalent and often more practical alternative to fresh cells for CAR-T manufacturing. Their reliable performance, coupled with the logistical advantages they confer, supports their adoption in both research and clinical settings to enhance the flexibility and accessibility of advanced cell therapies.
The functional state of T cells used in Chimeric Antigen Receptor (CAR) T-cell therapy directly determines therapeutic success. The phenotypic characteristics and exhaustion marker profiles of the final infusion product serve as key predictors of in vivo expansion, persistence, and ultimate clinical outcomes [64] [65]. As CAR-T therapy evolves, a central question has emerged: does the choice of starting material—fresh or cryopreserved peripheral blood mononuclear cells (PBMCs)—significantly alter these critical quality attributes of the final product? This comparison guide synthesizes current experimental data to address this question, providing researchers with objective, flow cytometry-based evidence to inform manufacturing decisions.
Evidence increasingly indicates that T-cells collected prior to extensive chemotherapy exposure retain superior fitness characteristics, supporting the strategy of early cell banking for autologous therapy or the use of healthy donor cells for allogeneic approaches [29] [66]. This analysis directly compares the phenotypic outcomes of CAR-T products manufactured via both fresh and cryopreserved pathways.
Comparative Data Analysis: Exhaustion Markers and Phenotypic Profiles
Exhaustion Marker Expression in Final CAR-T Products
Table 1: Comparison of Key Exhaustion Markers in CAR-T Final Products
| Exhaustion Marker | Association with Clinical Outcomes | Comparative Expression in Fresh vs. Cryopreserved-Derived CAR-T | Key Supporting Studies |
|---|---|---|---|
| LAG-3 | Higher expression linked to early relapse and MRD-positive outcomes [64] | No significant differences reported [4] [7] | Finney et al., Tao et al. [64] |
| TIM-3 | Co-expression with LAG-3 associated with dysfunctional CD8+ CAR-T populations [64] | Comparable levels observed between manufacturing approaches [4] | CD22 CAR-T trial analysis [65] |
| PD-1 | Transient expression is normal; persistent high co-expression with other markers defines severe exhaustion [64] | Consistent expression profiles across products from both starting materials [7] | Fraietta et al., Deng et al. [64] |
| TIGIT | Elevated levels correlate with reduced persistence and effector function [67] | Similar patterns observed in products from both fresh and cryopreserved PBMCs | Systematic CRISPR screens [67] |
T-cell Differentiation Phenotypes
Table 2: T-cell Differentiation States in CAR-T Manufacturing
| Cell Phenotype | Functional Significance | Impact of Cryopreservation on Frequency | Clinical Correlation |
|---|---|---|---|
| Naïve T-cells (TN) | Enhanced persistence, stem-like properties, proliferative capacity [4] | Proportions remain stable post-cryopreservation [4] | Associated with durable remissions and long-term persistence [65] |
| Central Memory T-cells (TCM) | Strong expansion upon antigen encounter, longevity [4] | No significant loss during frozen storage [4] [3] | Predictive of response in CLL and DLBCL [65] |
| Effector Memory T-cells (TEM) | Immediate cytotoxicity, potentially shorter persistence | Unchanged differentiation patterns during manufacturing [7] | High proportions may correlate with initial efficacy but rapid contraction |
| Terminally Differentiated | Limited replicative potential, senescent features | Not increased in cryopreserved-derived products [4] | Poor expansion and persistence post-infusion |
Experimental Protocols for Comparative Phenotype Analysis
Standardized Flow Cytometry Panel for CAR-T Product Characterization
Comprehensive immunophenotyping requires multiparameter flow cytometry panels capable of capturing differentiation, activation, and exhaustion states simultaneously. The following protocol is adapted from methodologies used in multiple comparative studies [4] [29] [7].
Sample Preparation:
- Staining Buffer: MACS buffer with 0.5% BSA (Miltenyi Biotec)
- Viability Stain: 1X Live/Dead Fixable Yellow Cell Stain (Invitrogen, ThermoFisher Scientific), 30 minutes at room temperature [7]
- FC Block: Human FC block (BD Biosciences), 30 minutes at room temperature
- Surface Staining: Antibody cocktail, 30 minutes at 4°C
- CAR Detection: Anti-FMC63 antibody for CD19-CARs (Acrobiosystems) [7]
Antibody Panel Design:
- Differentiation Panel: CD45RO, CCR7, CD62L, CD27, CD95
- Exhaustion Panel: PD-1, LAG-3, TIM-3, TIGIT, CD39, CD38
- Activation Panel: CD25, CD69, CD137 (4-1BB), HLA-DR
- Subset Panel: CD3, CD4, CD8, CD19 (for trogocytosis detection)
Instrumentation and Analysis:
- Flow Cytometer: CytoFLEX LX (Beckman Coulter) or equivalent high-parameter system
- Analysis Software: CytExpert (Beckman Coulter), FlowJo (BD)
- Gating Strategy: Lymphocyte gate → single cells → live cells → CD3+ T-cells → CD4+/CD8+ subsets → phenotypic markers [29]
Functional Validation Assays
Repetitive Stimulation Assay:
- Co-culture CAR-T cells with target cells at 1:1 effector-to-target ratio
- Re-stimulate every 3 days with fresh target cells for 3-5 cycles
- Monitor cytokine production (IFN-γ, TNF-α, IL-2) and activation markers after each stimulation [29]
Cytotoxicity Assays:
- Real-Time Cell Analysis (RTCA): Measure killing of tumor cell lines (e.g., SKOV-3, OCI-LY3) at multiple E:T ratios (e.g., 4:1, 2:1)
- Endpoint Measurements: Flow cytometry-based killing using count beads or impedance-based systems [4]
Cytokine Release Profiling:
- Multiplex cytokine arrays (IFN-γ, IL-2, IL-6, IL-10, TNF-α) measured 24 hours after co-culture with target cells
- No systematic differences in cytokine profiles between fresh and cryopreserved-derived CAR-T products observed [4] [7]
Technical Diagrams and Workflows
Comparative Manufacturing and Analysis Workflow
T-cell Exhaustion and Differentiation Pathways
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for CAR-T Phenotype Analysis
| Reagent Category | Specific Products | Research Application | Considerations |
|---|---|---|---|
| Cryopreservation Media | CryoStor CS10 (BioLife Solutions) | Maintains PBMC viability and function during frozen storage | 10% DMSO concentration; controlled-rate freezing critical [3] |
| T-cell Activation | Dynabeads Human T-Activator CD3/CD28 (ThermoFisher) | Standardized T-cell activation pre-transduction | Bead:cell ratio optimization required (typically 1:3) [29] |
| Cell Culture Cytokines | IL-2, IL-7, IL-15 (Miltenyi, PeproTech) | Support T-cell expansion and memory formation | IL-7/IL-15 favor stem-like memory phenotypes [29] |
| Flow Cytometry Antibodies | Anti-human PD-1, LAG-3, TIM-3, TIGIT (BioLegend, BD) | Exhaustion marker profiling | Combinatorial panels more informative than single markers [64] |
| Viability Stains | Zombie UV Fixable Viability Kit (BioLegend) | Exclusion of dead cells in flow analysis | Superior to PI/7-AAD for fixed samples [29] |
| CAR Detection Reagents | Anti-FMC63 Idiotype Antibodies (Acrobiosystems) | Transduction efficiency measurement | Critical for normalizing functional assays by CAR+ cell number [7] |
| Magnetic Separation | EasySep Human T Cell Isolation Kit (STEMCELL) | T-cell enrichment from PBMCs | Negative selection preserves T-cell activation potential [29] |
The comprehensive analysis of phenotypic and exhaustion markers in CAR-T products reveals a consistent theme: cryopreservation of starting PBMCs does not fundamentally alter the critical quality attributes of the final cellular product when compared to fresh PBMC processing [4] [3] [7]. While minor variations in expansion kinetics may occur during manufacturing, the functional potency, differentiation status, and exhaustion profiles remain comparable across both approaches.
This evidence supports the strategic implementation of "collect-and-freeze" models in CAR-T development and manufacturing [66]. The ability to bank PBMCs from patients during earlier disease stages or from healthy donors provides manufacturing flexibility and potentially access to higher-quality T-cells that have not been compromised by extensive prior therapies [29]. For researchers and developers, this means that logistical considerations—rather than concerns about product quality—can drive decisions regarding fresh versus cryopreserved starting materials.
The integration of multiparameter flow cytometry panels focusing on both differentiation (CD45RO, CCR7) and exhaustion markers (LAG-3, TIM-3, PD-1) provides critical insights that can inform manufacturing process development and potentially predict clinical performance [64] [65]. As the field advances, these phenotypic analyses will continue to guide the optimization of next-generation CAR-T therapies with enhanced efficacy and persistence.
The convergence of HIV and cancer management presents a complex clinical challenge, particularly for patients with AIDS-related diffuse large B-cell lymphoma (AR-DLBCL). These patients require simultaneous administration of antiretroviral therapy (ART) and chemotherapy, raising concerns about potential drug-drug interactions and compounded toxicities. Second-generation integrase strand transfer inhibitors (INSTIs), including bictegravir and dolutegravir, have become cornerstone treatments for HIV due to their favorable efficacy and tolerability profiles. However, their safety profile when combined with cytotoxic chemotherapy remains incompletely characterized in real-world clinical settings. This single-center retrospective analysis aims to bridge this knowledge gap by evaluating the safety and efficacy of pharmacotherapy combining second-generation INSTIs with chemotherapy in patients with AR-DLBCL.
Methodology
Study Design and Patient Population
This retrospective cohort study analyzed 96 newly diagnosed AR-DLBCL patients treated at the Public Health Clinical Center of Chengdu between February 2020 and May 2023 [68]. All enrolled patients received a second-generation INSTI-based antiretroviral regimen concurrently with chemotherapy. The study implemented comprehensive monitoring protocols throughout the treatment course.
Key Inclusion Criteria:
- Newly diagnosed AR-DLBCL
- Receiving second-generation INSTI-based ART
- Undergoing chemotherapy with R±CHOP or R±EPOCH regimens
Treatment Regimens
Patients received one of two antiretroviral therapy combinations alongside their chemotherapy [68]:
| ART Component | Regimen 1 | Regimen 2 |
|---|---|---|
| INSTI | Bictegravir | Dolutegravir |
| NRTI Backbone | Tenofovir alafenamide/emtricitabine | Lamivudine/albuvirtide |
| Patient Count | 60 | 36 |
Chemotherapy regimens consisted of either R±CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone) administered to 75 patients, or R±EPOCH (rituximab, etoposide, doxorubicin, vincristine, cyclophosphamide, prednisone) given to 21 patients [68].
Assessment Criteria
Primary Endpoints:
- Frequency and severity of adverse effects (AEs) assessed using Common Terminology Criteria for Adverse Events (CTCAE) version 4.02 [68]
Secondary Endpoints:
- CD4 count and CD4/CD8 ratio
- HIV viral load
- Complete response (CR), partial response (PR), and overall response rate (ORR) at end of treatment [68]
Evaluations were performed at each chemotherapy cycle with a median follow-up of 15.5 months (range: 5-33 months) [68].
Results
Safety Outcomes
The combination of second-generation INSTIs with chemotherapy demonstrated a manageable safety profile. The most common grade 3 or higher adverse events were hematologic toxicities [68]:
Table: Grade 3+ Adverse Events
| Adverse Event | Frequency (%) |
|---|---|
| Neutropenia | 32.29% |
| Thrombocytopenia | 20.83% |
Seven patients experienced serious complications during treatment, including pulmonary tuberculosis (2 cases), multiple organ dysfunction (1 case), intracranial infection (1 case), renal failure (1 case), and severe COVID-19 (2 cases). These complications resulted in 3 deaths [68]. No unexpected drug-drug interactions were reported, and viral load rebound was not observed throughout the study period [68].
Immunologic and Virologic Parameters
CD4 count and CD4/CD8 ratio showed only slight decreases from baseline to the sixth month of treatment [68]:
Table: Immunologic Parameters
| Parameter | Baseline | 6 Months | P-value |
|---|---|---|---|
| CD4 count (cells/μL) | 251.76 ± 188.53 | 233.44 ± 140.53 | 0.375 |
| CD4/CD8 ratio | 0.71 ± 0.69 | 0.66 ± 0.55 | 0.608 |
The stability of these immunologic parameters demonstrates that the combination therapy did not significantly compromise immune recovery despite concurrent chemotherapy [68].
Treatment Efficacy
The pharmacotherapy combination demonstrated substantial efficacy against AR-DLBCL [68]:
Table: Efficacy Outcomes
| Efficacy Parameter | Rate (%) |
|---|---|
| Objective Response Rate (ORR) | 85.41% |
| Complete Response (CR) Rate | 51.04% |
As of June 2024, 15 patients had died from severe infections or progressive disease. The overall response data indicate that second-generation INSTIs can be effectively combined with chemotherapy without compromising antitumor efficacy [68].
Discussion
Clinical Implications
This analysis provides reassuring evidence that second-generation INSTIs represent a safe and effective antiretroviral option for AR-DLBCL patients undergoing chemotherapy. The preservation of virologic suppression throughout treatment is particularly noteworthy, as uncontrolled HIV replication could further complicate cancer management. The hematologic toxicities observed were consistent with expected chemotherapy side effects rather than exacerbated toxicity from drug interactions.
Methodological Considerations
The retrospective design and single-center nature of this analysis limit generalizability. The relatively small sample size (n=96) may have underpowered the detection of rare adverse events. Additionally, the non-randomized treatment allocation introduces potential confounding factors. Future prospective, multi-center studies with larger cohorts would strengthen these findings.
This retrospective analysis demonstrates that second-generation INSTIs can be safely combined with chemotherapy in AR-DLBCL patients, without compromising antiretroviral efficacy or chemotherapy tolerability. The observed safety profile and high response rates support using these INSTIs as first-line antiretroviral therapy for HIV-positive patients requiring chemotherapy for lymphoma, regardless of the chemotherapy regimen selected.
Experimental Protocols & Methodologies
Study Design Framework
The following diagram illustrates the overall structure of this retrospective analysis:
Patient Management and Monitoring Protocol
The clinical management of patients in this study followed a structured monitoring protocol:
The Scientist's Toolkit: Research Reagent Solutions
Table: Key Reagents and Research Tools
| Reagent/Assay | Function/Application | Specification |
|---|---|---|
| Second-generation INSTIs | HIV integrase inhibition | Bictegravir/Tenofovir alafenamide/Emtricitabine; Dolutegravir/Lamivudine/Albuvirtide [68] |
| Chemotherapy Agents | Cytotoxic tumor cell killing | Rituximab, Cyclophosphamide, Doxorubicin, Vincristine, Prednisone, Etoposide [68] |
| CTCAE v4.02 | Standardized toxicity grading | Common Terminology Criteria for Adverse Events version 4.02 [68] |
| Flow Cytometry | Immune cell quantification | CD4+ T-cell count, CD4/CD8 ratio monitoring [68] |
| HIV Viral Load Assay | Virologic suppression monitoring | PCR-based quantification of HIV RNA [68] |
| Lugano Classification | Response assessment criteria | Standardized lymphoma response evaluation [68] |
The manufacturing of chimeric antigen receptor T-cell (CAR-T) therapies stands at a pivotal crossroads, where the transition from research to clinical application demands robust, scalable, and reproducible processes. The choice of starting material—particularly fresh versus cryopreserved peripheral blood mononuclear cells (PBMCs)—represents a fundamental decision that significantly influences manufacturing success across diverse engineering platforms [4]. This comprehensive analysis objectively evaluates the performance of cryopreserved PBMCs across three prominent CAR-T manufacturing systems: lentiviral transduction, non-viral transposon systems (specifically PiggyBac), and rapid ("Fast") CAR-T production platforms.
Current CAR-T production faces a paradoxical dilemma: while a significant proportion of patients are unable to receive treatment due to leukapheresis failure or rapid disease progression, the field still critically depends on cells sourced from these same immunocompromised patients [3]. Cryopreservation of PBMCs or leukapheresis products offers a strategic solution by decoupling cell collection from manufacturing, thereby introducing unprecedented flexibility into the therapeutic pipeline [5]. This approach enables advance collection from patients during healthier stages or from healthy donors, potentially mitigating issues of T-cell dysfunction associated with advanced disease and prior chemotherapy [4].
The validation of cryopreserved starting materials across multiple manufacturing platforms is not merely an academic exercise but an industrial necessity. As CAR-T therapies expand beyond hematologic malignancies into autoimmune diseases and solid tumors, establishing platform-agnostic starting materials becomes crucial for scalable, distributed manufacturing models [69] [3]. This guide synthesizes experimental data from recent studies to provide researchers, scientists, and drug development professionals with evidence-based comparisons of cryopreserved PBMC performance across the leading CAR-T manufacturing technologies.
Experimental Protocols and Methodologies
Cryopreservation and Thawing Protocols
The foundational methodology across all compared studies involves standardized cryopreservation and thawing procedures optimized for CAR-T manufacturing:
Cryoprotectant Formulation: Studies utilized clinical-grade cryoprotectants containing 10% DMSO (CS10), with strict control of DMSO concentration to ensure consistent cryoprotection efficacy [3]. The cryomedium maintained ≥7.5% DMSO concentration despite residual volume retention (3ml per 1×10^9 cells).
Controlled-Rate Freezing: Implementation of standardized freezing protocols using controlled-rate freezers (e.g., Thermo Profile 4 system) with the interval from cryoprotectant addition to freezing initiation strictly limited to ≤120 minutes to prevent ice crystal formation [3]. Target cell concentration was optimized at 5×10^7–8×10^7 cells/ml with formulation volumes of 20ml/bag.
Thawing and Recovery: Rapid thawing in water baths at 37°C followed by immediate processing. Post-thaw viability thresholds were established at ≥90% as a critical quality attribute (CQA), with viability measurements conducted via flow cytometry using annexin V/7-AAD or similar viability stains [4] [3].
Platform-Specific Manufacturing Protocols
Non-Viral PiggyBac Transposon System
The PiggyBac electroporation protocol was systematically optimized for cryopreserved PBMCs [4]:
- Cell Preparation: Cryopreserved PBMCs were thawed and enriched for T-cells using CD4/CD8 magnetic bead separation after resting.
- Activation: Cells were activated for 48 hours using anti-CD3/CD28 activators prior to electroporation.
- Electroporation Parameters: The mesothelin (MSLN) CAR vector was delivered via electroporation using optimized voltage and waveform parameters specifically adjusted for post-thaw cell characteristics.
- Expansion Culture: Transfected cells were cultured for 11 days in optimized media containing IL-2, with process optimizations focusing on enhancing proliferation and reducing exhaustion markers.
Lentiviral Transduction System
The lentiviral CAR-T manufacturing process was adapted for cryopreserved starting materials [3] [5]:
- Cell Recovery: Cryopreserved leukapheresis products were thawed and underwent centrifugation-based impurity removal to eliminate residual red blood cells and platelets.
- Transduction Protocol: T-cell activation was followed by transduction with lentiviral vectors encoding anti-CD19 CAR at optimized multiplicity of infection (MOI).
- Expansion: Transduced cells were expanded over 14 days with regular feeding and cytokine supplementation (IL-2, IL-15).
- Quality Monitoring: In-process controls included flow cytometry for CAR expression, cell counting, and viability assessment.
Fast CAR-T System
The rapid manufacturing platform emphasized abbreviated culture periods [57] [3]:
- Accelerated Activation: Intensive activation protocols reduced the activation phase to 24 hours.
- Abbreviated Culture: Total manufacturing time was compressed to 7-9 days through optimized media formulations and enhanced transduction efficiency.
- Process Intensification: Higher cell densities and integrated processing steps minimized hands-on time and overall vein-to-vein time.
Analytical and Functional Assays
Standardized assays were implemented across studies to enable cross-platform comparisons:
Phenotypic Characterization: Multicolor flow cytometry panels analyzing differentiation markers (CD45RO, CCR7, CD62L, CD95) to determine T-cell subsets (naïve, stem cell memory, central memory, effector memory, terminal effector) [4] [3] [70].
Functional Assessments:
- Cytotoxicity: Real-time cellular analysis (RTCA) against target tumor cells (e.g., SKOV-3 ovarian cancer cells) at effector-to-target ratios of 2:1 and 4:1 [4].
- Cytokine Secretion: Multiplex cytokine profiling (IFN-γ, IL-2, IL-4, IL-5, IL-6, IL-10, IL-13, TNF-α) via ELISA or Luminex [4] [5].
- Exhaustion Markers: PD-1, LAG-3, TIM-3 expression analysis by flow cytometry [4].
Expansion Capacity: Cumulative population doublings and fold expansion calculations throughout culture periods [4] [3].
Comparative Performance Across Manufacturing Platforms
Table 1: Comprehensive comparison of cryopreserved PBMC performance across CAR-T manufacturing platforms
| Performance Metric | Non-Viral (PiggyBac) | Lentiviral | Fast CAR-T |
|---|---|---|---|
| Post-Thaw Viability | 90.95% (3.5 years cryopreservation) [4] | 90.9-97.0% [3] | ≥90% [3] |
| Transduction Efficiency | Comparable to fresh [4] | 42.01-51.21% CD3+ purity [3] | Comparable to fresh [3] |
| Fold Expansion | Slight reductions (not significant) [4] | Comparable to fresh [3] | Comparable to fresh leukapheresis [3] |
| T-cell Subsets (Tn/Tcm) | No significant difference vs. fresh at harvest [4] | Preserved phenotypic profiles [3] | Maintained with optimized protocols [3] |
| Exhaustion Markers | No significant difference vs. fresh [4] | Comparable expression profiles [3] | Not specifically reported |
| Cytotoxicity (%) | 91.02-100% (E:T 4:1) [4] | Comparable to fresh [3] | Comparable to fresh [3] |
| Cytokine Secretion | Comparable (except IFN-γ decrease in CAR-12M) [4] | No significant difference in cytokine profiles [5] | Not specifically reported |
| Clinical Response | N/A (preclinical) | 46.2% CR, 69.2% ORR (DLBCL) [5] | Similar to standard manufacturing [3] |
Platform-Specific Performance Analysis
Non-Viral (PiggyBac) System Performance
The PiggyBac transposon system demonstrates particular compatibility with cryopreserved PBMCs, overcoming historical challenges with non-viral transfection of frozen cells [4]. Critical findings include:
Long-Term Stability: PBMCs cryopreserved for up to 3.5 years maintained high viability (90.95%), enabling successful manufacturing without significant timeline-dependent degradation [4].
Phenotypic Preservation: CAR-T products generated from cryopreserved PBMCs exhibited comparable differentiation profiles to fresh counterparts, with no significant differences in naïve (Tn) and central memory (Tcm) T-cell proportions at harvest—a critical determinant of in vivo persistence and efficacy [4] [70].
Functional Competence: Despite a noted decrease in IFN-γ secretion in cells derived from 12-month cryopreserved PBMCs, cytotoxicity remained uncompromised across all timepoints, demonstrating dissociation between specific cytokine secretion patterns and effector function in this context [4].
Lentiviral System Performance
Lentiviral transduction, as the most established manufacturing platform, shows robust compatibility with cryopreserved starting materials:
Clinical Validation: A retrospective study of 162 relapsed/refractory DLBCL patients demonstrated no significant differences in key clinical efficacy metrics between cryopreserved and fresh PBMC-derived CAR-T products, including overall survival (75.4% vs. 64.1% at 1 year) and progression-free survival (52.1% vs. 44.5%) [5].
Pharmacokinetic Equivalence: Critical metrics of CAR-T cell expansion and persistence revealed no significant differences in achievement of target infusion dose (46.1% vs. 44.9%), in vivo persistence (median 21 days for both), or peak CAR-T cell proportions in lymphocytes [5].
Safety Profile Consistency: The incidence and severity of cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) were equivalent between groups, alongside comparable peak levels of inflammatory cytokines including IL-6, IL-10, and IFN-γ [5].
Fast CAR-T System Performance
Rapid manufacturing platforms maintain critical quality attributes with cryopreserved starting materials while significantly reducing vein-to-vein time:
Accelerated Timelines: Cryopreservation compatibility enables manufacturing timelines compressed to 7-9 days, addressing the critical need for rapid access in patients with aggressive disease [57] [3].
Process Flexibility: The decoupling of cell collection from manufacturing through cryopreservation facilitates seamless integration with rapid production platforms, potentially reducing the 20% patient attrition between leukapheresis and infusion observed in conventional timelines [57].
Technical Workflow and Signaling Pathways
Multi-Platform Manufacturing Workflow
The following diagram illustrates the integrated experimental workflow for comparative assessment of cryopreserved PBMCs across CAR-T manufacturing platforms:
Diagram Title: Cryopreserved PBMC CAR-T Manufacturing Workflow
T-cell Differentiation Pathway
Understanding T-cell maturation is essential for interpreting phenotypic data in comparative studies:
Diagram Title: T-cell Differentiation and Phenotypic Markers
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key research reagent solutions for cryopreserved PBMC CAR-T manufacturing
| Reagent/Material | Function | Application Notes |
|---|---|---|
| CS10 Cryoprotectant | 10% DMSO formulation for cell preservation | Maintain ≥7.5% final DMSO concentration; strict ≤120min processing before freezing [3] |
| CD4/CD8 Magnetic Beads | T-cell enrichment from PBMCs | Critical for non-viral systems; compatible with post-thaw cells [4] |
| PiggyBac Transposon System | Non-viral gene delivery | Optimized electroporation parameters for cryopreserved cells; 100kb cargo capacity [4] |
| Lentiviral Vectors | Viral gene delivery | Consistent transduction efficiency with cryopreserved PBMCs; standard payload capacity [3] [5] |
| Anti-CD3/CD28 Activators | T-cell activation and expansion | 48-hour activation for standard systems; 24-hour for Fast CAR-T [4] [57] |
| Cytokines (IL-2, IL-7, IL-15) | T-cell survival and differentiation | Concentration optimization critical for maintaining Tcm phenotype [4] [70] |
| Viability Stains (7-AAD/Annexin V) | Apoptosis and viability assessment | Essential for post-thaw quality control; ≥90% viability threshold [4] [3] |
| Phenotypic Antibody Panels | T-cell subset characterization | Must include CD45RA, CD45RO, CCR7, CD62L, CD95 for differentiation staging [4] [70] |
Discussion and Industrial Implications
Strategic Advantages of Platform-Agnostic Cryopreserved Starting Materials
The consistent performance of cryopreserved PBMCs across diverse manufacturing platforms demonstrates their viability as universal starting materials for CAR-T production. This compatibility delivers several strategic advantages:
Supply Chain Resilience: Decoupling cell collection from manufacturing through cryopreservation mitigates risks associated with fresh material logistics, including time-sensitive viability decay (typically 24-72 hour transport windows for fresh leukapheresis) [3]. This flexibility is particularly valuable in the context of global clinical trials and distributed manufacturing models.
Manufacturing Flexibility: Cryopreservation enables advance collection from patients during healthier clinical stages or from healthy donors, potentially overcoming T-cell dysfunction associated with advanced disease and intensive prior therapies [4]. This approach directly addresses the challenge wherein nearly 30% of patients prescribed CAR-T therapy never undergo leukapheresis, and 20% of those who do fail to reach infusion [57].
Quality Standardization: Batch-based manufacturing from cryopreserved stocks facilitates quality control testing before CAR-T production, potentially reducing lot-to-lot variability and manufacturing failures [3]. Standardized cryopreserved starting materials could improve comparability across clinical trials and commercial products.
Limitations and Research Gaps
Despite the compelling evidence supporting cryopreserved PBMCs, several limitations merit consideration:
Process Standardization: While individual studies demonstrate optimized protocols, the field lacks universally standardized cryopreservation procedures, particularly for leukapheresis products which remain understudied (representing only 18.3% of 349 studies from 2010-2024) [3].
Long-Term Functional Data: Most clinical validation studies report intermediate endpoints (e.g., 1-year survival); longer-term follow-up is needed to fully establish equivalence in durable responses, particularly given the importance of T-cell persistence for sustained efficacy [5].
Solid Tumor Applications: The current clinical validation predominantly concerns hematologic malignancies (particularly DLBCL); expanded investigation is warranted in solid tumor settings where the tumor microenvironment presents distinct challenges to T-cell function [71].
This multi-platform validation demonstrates that cryopreserved PBMCs constitute a viable and robust starting material across leading CAR-T manufacturing systems, performing comparably to fresh cells in critical quality attributes including expansion capacity, phenotypic profiles, and functional potency. The experimental evidence supports cryopreservation as a strategic enabler for more flexible, scalable, and resilient CAR-T manufacturing paradigms.
The consistency of results across lentiviral, non-viral PiggyBac, and Fast CAR-T systems suggests that cryopreservation compatibility is a platform-agnostic feature, potentially simplifying process development and technology transfer across manufacturing sites. As CAR-T therapy continues to expand into new disease indications and geographic markets, the adoption of cryopreserved starting materials will play a pivotal role in overcoming current limitations in manufacturing scalability and accessibility.
Future research directions should focus on further standardization of cryopreservation protocols, validation in emerging manufacturing platforms (including allogeneic approaches), and extended clinical follow-up to confirm long-term equivalence. The integration of cryopreserved PBMCs into mainstream CAR-T manufacturing represents a critical step toward realizing the full potential of cell therapy across diverse patient populations and clinical settings.
Chimeric antigen receptor T-cell (CAR-T) therapy represents a revolutionary advance in cancer treatment, yet its manufacturing process faces significant logistical and economic challenges. Traditional reliance on fresh peripheral blood mononuclear cells (PBMCs) from patients creates complex supply chain vulnerabilities and manufacturing constraints. This comparison guide objectively evaluates the alternative approach of using cryopreserved PBMCs, examining both logistical advantages and functional performance across key manufacturing parameters.
The conventional model utilizing fresh patient cells encounters numerous obstacles, including manufacturing failures and logistical hurdles associated with coordinating cell collection, transport, and processing within constrained timelines [4]. Cryopreserved PBMCs offer a potential solution by decoupling cell collection from manufacturing, thereby introducing flexibility into the supply chain. While lentiviral processes for generating CAR-T from cryopreserved cells have been reported, successful methods using the PiggyBac electroporation system have been less documented [4] [58].
This guide systematically compares fresh versus cryopreserved PBMCs for CAR-T manufacturing, presenting experimental data on cell viability, phenotypic stability, expansion potential, and cytotoxic functionality to inform research and development decisions.
Experimental Protocols and Methodologies
PBMC Collection and Cryopreservation Protocols
The integrity of PBMC samples begins with standardized collection procedures. According to gold-standard protocols developed by the Office of HIV/AIDS Network Coordination (HANC), several critical factors must be controlled:
- Anticoagulant Selection: Blood collection should use clinically-convenient anticoagulant-lined vacuum-tubes, with documentation of whether EDTA, heparin, or citrate was used [25].
- Processing Time and Temperature: The HANC-SOP recommends that processing time should not exceed 8 hours at controlled temperatures [25]. Processing delays of 24 hours or more have been associated with reduced cell viability [25].
- Isolation Method: PBMCs are typically isolated using density-gradient centrifugation methods (e.g., Ficoll-Paque) or cell preparation tubes (CPTs) [25]. The isolation method and processing technician should be documented as technician experience contributes significantly to variability in cell recovery [25].
For cryopreservation, the HANC-SOP specifies that PBMCs should be gently resuspended to 10⁷/mL in a cryopreservation medium containing 10% DMSO and 90% Foetal Calf Serum (FCS), cooled to 2-8°C with continuous swirling [25]. Long-term storage should maintain temperatures below -132°C (the glass transition temperature of water) to preserve cell viability and function [72].
CAR-T Generation and Evaluation Methods
Recent studies have established standardized protocols for generating CAR-T cells from cryopreserved PBMCs using non-viral transposon systems:
- Cell Source and Preparation: PBMCs are obtained from healthy donors and divided into fresh and cryopreserved cohorts. Cryopreserved samples are stored for varying durations (3 months to 2 years) in liquid nitrogen before analysis [4].
- CAR-T Manufacturing: After thawing, T-cells are enriched using CD4/CD8 magnetic beads and activated for 48 hours. The mesothelin (MSLN) CAR vector is then introduced via the PiggyBac electroporation system [4].
- Functional Assessment: CAR-T cells are evaluated through expansion tracking, phenotypic characterization (flow cytometry for differentiation and exhaustion markers), and functional assays including cytotoxicity tests and cytokine release measurements [4].
Table 1: Key Experimental Parameters in Comparative Studies
| Parameter | Fresh PBMCs | Cryopreserved PBMCs | Assessment Method |
|---|---|---|---|
| Initial Viability | >95% [73] | 90-95% [4] [73] | Trypan blue exclusion |
| Processing Timeline | Immediate processing | Flexible processing | HANC-SOP guidelines [25] |
| T-cell Proportion | Stable baseline | Stable post-thaw [4] | Flow cytometry |
| CAR Transfection Efficiency | Platform-dependent | Comparable to fresh [4] | Flow cytometry for CAR expression |
| Cryopreservation Medium | N/A | 10% DMSO in FCS or serum-free alternatives [25] [31] | Multiple viability assessments |
Figure 1: Comparative Experimental Workflow for Fresh vs. Cryopreserved PBMCs in CAR-T Manufacturing
Comparative Performance Analysis
Impact of Cryopreservation on PBMC Viability and Phenotype
Long-term cryopreservation maintains PBMC viability at functionally relevant levels, though a slight reduction compared to fresh samples is observable. Studies evaluating different cryopreservation durations—3 months (3M), 6 months (6M), 12 months (12M), and 2 years (2Y)—revealed that although there was a statistically significant difference in cell viability between fresh PBMCs and cryopreserved ones, the actual decrease was only 4.00% to 5.67% [4]. Moreover, viability remained stable regardless of freezing duration, with samples frozen for 3.5 years showing average viability of 90.95% [4].
Phenotypic analysis reveals critical maintenance of key cell populations:
- T-cell Proportions: The proportion of T-cells remains relatively stable following cryopreservation, indicating that CAR-T preparation is largely unaffected as it primarily derives from CD3+ T cells [4].
- Naïve and Memory T-cells: T-cell differentiation states were investigated by staining for CD45RO and CCR7, revealing no significant changes in naïve Tn (CD45RO-CCR7+) and central memory Tcm (CD45RO+CCR7+) proportions post-cryopreservation compared to fresh samples [4]. These subsets are crucial for enhancing CAR-T activation, persistence, and effector function [4].
- NK and B-cells: Flow cytometry analysis revealed a decrease in the proportions of natural killer (NK) cells and B cells following cryopreservation, attributed to their greater sensitivity to hypothermic conditions [4].
Table 2: Viability and Phenotypic Stability of Cryopreserved vs. Fresh PBMCs
| Parameter | Fresh PBMCs | Cryopreserved (3M-2Y) | Significance |
|---|---|---|---|
| Viability | >95% [73] | 90-95% [4] | Slight decrease (4-6%) but functionally stable |
| T-cell Proportion | Baseline | Stable [4] | No significant impact on CAR-T potential |
| Tn/Tcm Populations | Baseline | Maintained [4] | Critical for long-term persistence |
| NK/B-cells | Baseline | Decreased [4] | Minimal impact on CAR-T generation |
| Recovery Rate | N/A | ≥90% with optimized media [31] | Dependent on cryopreservation protocol |
CAR-T Functional Performance Comparison
CAR-T cells generated from cryopreserved PBMCs demonstrate comparable functionality to those derived from fresh cells across critical performance parameters:
- Expansion Potential: CAR-T cells from cryopreserved PBMCs showed only slight reductions in proliferation across the 6M, 12M, and 2Y timelines, with no statistically significant impact on overall expansion potential [4].
- Phenotype and Differentiation: Throughout the culture process, assessments revealed consistent mean CD3+ purity, CD4+/CD8+ ratios, and transfection efficiency between CAR-T products derived from fresh and cryopreserved PBMCs [4].
- Exhaustion Markers: T-cell exhaustion markers showed consistent patterns between fresh and cryopreserved samples, suggesting that cryopreservation does not compromise CAR-T persistence [4].
- Cytotoxic Activity: In cytotoxicity assays against human ovarian cancer cell line (SKOV-3) cells at effector-to-target ratios of 4:1, CAR-T cells from both fresh (CAR-F) and 2-year cryopreserved PBMCs (CAR-2Y) showed comparable cytotoxicity: 91.02%-100.00% and 95.46%-98.07%, respectively [4]. Similarly, no statistically significant difference in cytotoxicity was observed at 2:1 ratios [4].
- Cytokine Secretion: Analysis of cytokine secretion showed that IFN-γ had a significant decrease in CAR-12M compared to CAR-F, though cytotoxic function remained unaffected [4]. No systematic changes were observed in the secretion of other cytokines (IL-6, IL-10, IL-5, IL-4, IL-13, IL-2, TNF-α) due to PBMC cryopreservation [4].
Process Optimization and Advanced Cryopreservation Formats
Recent advances in cryopreservation methodologies have further enhanced the feasibility of using cryopreserved starting materials:
- Cryopreserved Leukapheresis: Recent studies have demonstrated that cryopreserved leukapheresis achieves ≥90% post-thaw viability, with recovery and phenotypic profiles comparable to PBMCs [73]. It exhibited a higher lymphocyte proportion than PBMCs (66.59% vs. 52.20%), correlating with enhanced CAR-T potential [73].
- Serum-Free Cryopreservation Media: Alternatives to traditional FBS-containing media have been validated for long-term PBMC cryopreservation. Studies show that PBMCs cryopreserved in serum-free media like CryoStor CS10 and NutriFreez D10 maintained high viability and functionality comparable to FBS-based media across 2-year storage periods [31].
- DMSO Concentration Optimization: While 10% DMSO remains the standard concentration for cryopreservation media, studies have evaluated lower concentrations [31]. Media with DMSO concentrations below 7.5% showed significant viability loss and were eliminated after initial assessments [31].
Figure 2: CAR-T Functional Assessment Parameters and Outcomes
Logistical and Economic Advantages Framework
Supply Chain Resilience and Manufacturing Flexibility
The implementation of cryopreserved PBMCs introduces significant logistical advantages that address critical vulnerabilities in the CAR-T manufacturing supply chain:
- Decoupled Collection and Manufacturing: Cryopreservation enables the separation of cell collection from manufacturing processes, allowing for strategic stockpiling of starting materials and creating buffer inventory to mitigate against collection disruptions [4] [73].
- Extended Storage Stability: With demonstrated stability of PBMCs for up to 2 years and beyond without significant functional decline, organizations can implement longer-term inventory planning and reduce the urgency of immediate processing [4] [31].
- Distributed Manufacturing Models: Frozen PBMCs can be transported to specialized manufacturing facilities, enabling a hub-and-spoke model where collection occurs at multiple sites while manufacturing is centralized in optimized facilities [73].
- Quality Control Integration: The flexibility afforded by cryopreservation allows for comprehensive quality testing of starting materials before manufacturing initiation, reducing batch failure risks [4].
Economic and Operational Advantages
The transition to cryopreserved PBMC-based manufacturing generates substantial economic benefits:
- Reduced Manufacturing Failures: By enabling quality verification of starting materials before CAR-T generation, cryopreservation reduces costly batch failures that occur when using fresh cells of variable quality [4].
- Infrastructure Optimization: Manufacturing can be scheduled based on facility capacity rather than patient availability, improving equipment utilization and reducing idle capacity [73].
- Donor Selection Flexibility: Cryopreservation enables the use of healthy donor cells instead of patient cells, mitigating issues affecting treatment efficacy such as suboptimal cell condition following illness or delays in cell preparation [4]. Healthy donors may have a greater proportion of CD4+ T cells than patients with ALL, and have a greater capacity for expansion during CAR-T culture [4].
- Cost Reduction: The PiggyBac transposon system used with cryopreserved PBMCs overcomes limitations of viral transduction systems. Plasmids are easier to prepare and do not require viruses, which greatly reduces the cost of CAR-T preparation [4].
Table 3: Logistical and Economic Comparison of Fresh vs. Cryopreserved PBMC Platforms
| Parameter | Fresh PBMCs | Cryopreserved PBMCs | Advantage Impact |
|---|---|---|---|
| Manufacturing Timeline | Fixed (immediate processing) | Flexible scheduling | 30-50% reduction in scheduling constraints [74] |
| Supply Chain Complexity | High (time-sensitive transport) | Reduced (frozen transport) | Improved resilience to disruptions [75] |
| Facility Utilization | Suboptimal (patient-dependent) | Optimized (scheduled batches) | Increased throughput [73] |
| Starting Material Quality | Variable (patient-dependent) | Controllable (pre-screened) | Reduced batch failures [4] |
| Production Cost | Higher (viral systems, urgent processing) | Lower (non-viral options, planned processing) | Significant cost reduction potential [4] |
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Key Research Reagent Solutions for PBMC Cryopreservation and CAR-T Manufacturing
| Reagent/Category | Function/Purpose | Examples/Specifications |
|---|---|---|
| Cryopreservation Media | Cell protection during freezing | CryoStor CS10, NutriFreez D10, FBS+10% DMSO [31] |
| Cell Separation Media | PBMC isolation from whole blood | Ficoll-Paque, Lymphoprep, Cell Preparation Tubes (CPTs) [25] |
| Cell Activation Reagents | T-cell activation pre-transfection | CD3/CD28 activators, various cytokine combinations |
| Transfection Systems | CAR gene introduction | PiggyBac transposon system, Lentiviral systems [4] |
| Cell Culture Media | CAR-T expansion | Serum-free media, cytokine-supplemented formulations |
| Viability Assays | Quality assessment pre/post-cryopreservation | Trypan blue exclusion, flow cytometry with viability dyes [4] |
| Phenotyping Antibodies | Cell population characterization | CD3, CD4, CD8, CD45RO, CCR7 panels [4] |
| Functional Assay Kits | CAR-T potency assessment | Cytotoxicity assays, cytokine secretion measurements [4] |
The comprehensive comparison between fresh and cryopreserved PBMCs for CAR-T manufacturing reveals a compelling value proposition for cryopreserved platforms. While fresh PBMCs remain the traditional standard, cryopreserved alternatives demonstrate comparable functional performance in critical parameters including expansion potential, phenotypic characteristics, and cytotoxic activity.
The decisive advantage of cryopreserved PBMCs lies in their ability to enhance supply chain resilience and manufacturing flexibility. By decoupling cell collection from manufacturing processes, organizations can implement more robust and economically efficient production models. The logistical benefits—including reduced scheduling constraints, improved inventory management, and distributed manufacturing capabilities—address fundamental vulnerabilities in current CAR-T production systems.
For researchers and drug development professionals, the experimental evidence supports the adoption of cryopreserved PBMC platforms, particularly when integrated with optimized protocols and serum-free cryopreservation media. This approach not only maintains therapeutic quality but also introduces strategic advantages in scale-up and commercial translation of CAR-T therapies.
Conclusion
The collective evidence from foundational research, optimized methodologies, and clinical validation firmly establishes cryopreserved PBMCs and leukapheresis as a viable and often superior starting material for CAR-T manufacturing. Key takeaways confirm that while an initial, manageable reduction in viability post-thaw may occur, critical T-cell phenotypes, expansion potential, and the final cytotoxic function of the CAR-T product remain comparable to those derived from fresh cells. The logistical benefits—including decoupling manufacturing from complex fresh-cell logistics, enabling patient cell collection at a healthier stage, and facilitating scalable, distributed production models—are transformative. Future directions should focus on the large-scale clinical validation of these findings, further standardization and automation of cryopreservation protocols, and the integration of these strategies with next-generation, point-of-care manufacturing to truly democratize access to CAR-T therapy globally.
References
- 1. Why Separate Peripheral Blood Mononuclear Cells [https://www.akadeum.com/blog/pbmc-isolation-methods-isolation-white-blood-cells/?srsltid=AfmBOorOHj69qDzxqPc8R9DLlcAyL64zU-LmIN6wlq93F5LY1jtEteM_]
- 2. CAR-T cell manufacturing: Major process parameters and next ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10791545/]
- 3. a multi-platform comparative study | Scientific Reports [https://www.nature.com/articles/s41598-025-14865-5]
- 4. Comparative analysis and process optimization ... - Nature [https://www.nature.com/articles/s41598-025-89686-7]
- 5. Evaluating the Impact of Cryopreservation of PBMCs on ... [https://www.sciencedirect.com/science/article/pii/S0006497123101157]
- 6. CAR-T manufactured from frozen PBMC yield efficient ... [https://pubmed.ncbi.nlm.nih.gov/36225930/]
- 7. CAR-T manufactured from frozen PBMC yield efficient ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2022.1007042/full]
- 8. Impact of cryopreservation on CAR T production and clinical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9582437/]
- 9. Probing mechanisms of cryopreservation damage to ... [https://pubmed.ncbi.nlm.nih.gov/39918490/]
- 10. Impact of cryopreservation agents on sperm quality, DNA ... [https://www.nature.com/articles/s41598-025-15456-0]
- 11. Evaluation of the viability and functionality of human ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12355051/]
- 12. The effects of cryopreservation on PBMCs transcriptome ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1690316/full]
- 13. Effects of long-term cryopreservation of PBMC on recovery ... [https://www.sciencedirect.com/science/article/pii/S0022175921001265]
- 14. Cryopreserved PBMC Considerations | Immune System ... [https://sanguinebio.com/blog/cryopreserved-pbmc-considerations/?srsltid=AfmBOorsl118nzXr5aSOLAjn9fZBklYwok6bys51f-uJfdwRHrYU37m0]
- 15. Quality Assurance Program for Peripheral Blood ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2043300/]
- 16. a multi-platform comparative study - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12354897/]
- 17. Impact of cryopreservation on CAR T production and ... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2022.1024362/full]
- 18. A practical cryopreservation and staining protocol for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6060642/]
- 19. Protocol for Cryopreserving PBMCs [https://www.stemcell.com/how-to-cryopreserve-pbmcs.html]
- 20. Technical pitfalls when collecting, cryopreserving, thawing ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2024.1382192/full]
- 21. Impacts of cryopreservation on phenotype and functionality of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10956725/]
- 22. The effects of cryopreservation on PBMCs transcriptome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12575299/]
- 23. Optimizing the cryopreservation and post-thaw recovery of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10773738/]
- 24. Phenotypic and functional comparisons between ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12576105/]
- 25. Technical pitfalls when collecting, cryopreserving, thawing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11133553/]
- 26. The clinical outcomes of fresh versus cryopreserved CD19 ... [https://www.sciencedirect.com/science/article/abs/pii/S0011224020301462]
- 27. Flow Cytometric Immunophenotyping: Minimal Differences in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11505181/]
- 28. Understanding the freezing responses of T-cells and other ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7682753/]
- 29. Prior chemotherapy deteriorates T-cell quality for CAR T-cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11987159/]
- 30. Optimal Thawing of Cryopreserved Peripheral Blood ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3901099/]
- 31. Evaluation of the viability and functionality of human ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1627973/full]
- 32. Key quality parameter comparison of mesenchymal stem ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11324487/]
- 33. Evaluation of Nonviral piggyBac and lentiviral Vector in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8784881/]
- 34. Differences in the phenotypes and transcriptomic ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2023.1068625/full]
- 35. A guide in lentiviral vector production for hard-to-transfect ... [https://www.nature.com/articles/s41598-021-98657-7]
- 36. Optimization of lentiviral transduction parameters and its ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6464586/]
- 37. Optimization of metabolism to improve efficacy during CAR-T ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-021-03165-x]
- 38. Comparison of media for a human peripheral blood ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10655156/]
- 39. Use of serum-free media for peripheral blood mononuclear ... [https://www.frontiersin.org/journals/toxicology/articles/10.3389/ftox.2024.1462688/full]
- 40. Accelerating cell culture media development using ... [https://www.nature.com/articles/s41467-025-61113-5]
- 41. The impact of cryopreservation on cytokine secretion and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11491348/]
- 42. Buffy Coat vs Leukapheresis: Which Yields More PBMCs? [https://cgt.global/buffy-coat-vs-leukapheresis-choosing-the-right-cell-source-for-your-research/]
- 43. Flow Cytometric Immunophenotyping: Minimal Differences ... [https://pubmed.ncbi.nlm.nih.gov/39457632/]
- 44. Frequently Asked Questions on Primary Cells [https://www.stemcell.com/primary-cells-faq.html]
- 45. From cold chain to ambient: Benefits, risks, and evidence ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12593556/]
- 46. PBMC Storage - Cryopreservation & Controlled-Rate Freezing [https://researchdonors.co.uk/2025/02/18/the-importance-of-cryopreservation-solutions-and-controlled-rate-freezing-in-pbmc-storage/]
- 47. Cell Freezing and Preservation Media | Cell Storage [https://www.stemcell.com/cell-storage-media-overview.html]
- 48. Cell Freezing Media | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/cell-culture/mammalian-cell-culture/reagents/cell-freezing-media.html]
- 49. Choosing the Right Tool for Genetic Engineering: Clinical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8697565/]
- 50. Non-viral, high throughput genetic engineering of primary ... [https://www.sciencedirect.com/science/article/abs/pii/S014296122400615X]
- 51. Using Fresh vs. Cryopreserved PBMCs in Preclinical Research [https://cytologicsbio.com/should-you-order-fresh-or-cryopreserved-pbmcs/?srsltid=AfmBOooKpBEsJgtx_8LwlVZ4X6mg8tHWYW0dUFCNJ8cP-E8O_vj12dUS]
- 52. Physical non-viral gene delivery methods for tissue ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5102682/]
- 53. Comparison of two lab-scale protocols for enhanced ... [https://www.nature.com/articles/s41598-023-45197-x]
- 54. Optimized DNA electroporation for primary human T cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5789706/]
- 55. Keys to Successful Cell-Based Assay Development with ... [https://maxcyte.com/resource/keys-to-successful-cell-based-assay-development-with-scalable-electroporation/]
- 56. T-Cell Engineering For Cancer Immunotherapy [https://maxcyte.com/resource/efficient-and-scalable-t-cell-engineering-for-advancing-cancer-immunotherapy-of-hepatocellular-carcinoma-by-a-cgmp-compliant-non-viral-cell-engineering-platform/]
- 57. Full speed ahead: how rapid CAR-T manufacturing can ... [https://www.insights.bio/cell-and-gene-therapy-insights/journal/article/3513/full-speed-ahead-how-rapid-cart-manufacturing-can-shape-the-cell-therapy-landscape]
- 58. Comparative analysis and process optimization for ... [https://pubmed.ncbi.nlm.nih.gov/39934258/]
- 59. Frontiers | Epigenetic regulation of CD8+ T cell exhaustion [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1700039/full]
- 60. Proteotoxic stress response drives T cell exhaustion and ... [https://www.nature.com/articles/s41586-025-09539-1]
- 61. Optimizing PBMC Viability and Recovery: Updated Best ... [https://cgt.global/pbmc-viability-and-recovery/]
- 62. The effect of serum origin on cytokines induced killer cell ... [https://bmcimmunol.biomedcentral.com/articles/10.1186/s12865-023-00562-3]
- 63. Expansion With IL-15 Increases Cytotoxicity of Vγ9Vδ2 T ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7485111/]
- 64. The Phenotype of the Infused T cell Product Matters in ... [https://www.linkedin.com/pulse/phenotype-infused-cell-product-matters-clinic-part-3-arnaud-9fbgc]
- 65. Immunophenotype of CAR T cells and apheresis products ... [https://www.sciencedirect.com/science/article/abs/pii/S1525001625001935]
- 66. Manufacturing chimeric antigen receptor T cells from ... [https://www.sciencedirect.com/science/article/abs/pii/S1465324921007519]
- 67. Systematic discovery of CRISPR-boosted CAR T cell ... [https://www.nature.com/articles/s41586-025-09507-9]
- 68. Safety and Efficacy of Pharmacotherapy Containing the ... [https://pubmed.ncbi.nlm.nih.gov/40179836/]
- 69. In vivo chimeric antigen receptor (CAR)-T cell therapy | Nature Reviews Drug Discovery [https://www.nature.com/articles/s41573-025-01291-5]
- 70. New player in CAR-T manufacture field: comparison of ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1561174/full]
- 71. CAR T-Cell Therapy Market Size to Hit USD ... [https://www.precedenceresearch.com/car-t-cell-therapy-market]
- 72. The effects of storage temperature on PBMC gene expression [https://bmcimmunol.biomedcentral.com/articles/10.1186/s12865-016-0144-1]
- 73. leukapheresis enables scalable and distributed... Cryopreserved [https://pubmed.ncbi.nlm.nih.gov/40813416/]
- 74. Supply chains: To build resilience, manage proactively [https://www.mckinsey.com/capabilities/operations/our-insights/supply-chains-to-build-resilience-manage-proactively]
- 75. Supply Chain Resilience: Build an Agile, Future-Proof ... [https://www.inboundlogistics.com/articles/supply-chain-resilience-what-it-is-common-disruptions-and-how-to-build/]