mRNA Reprogramming in iPSC Generation: Mechanisms, Clinical Applications, and Future Directions
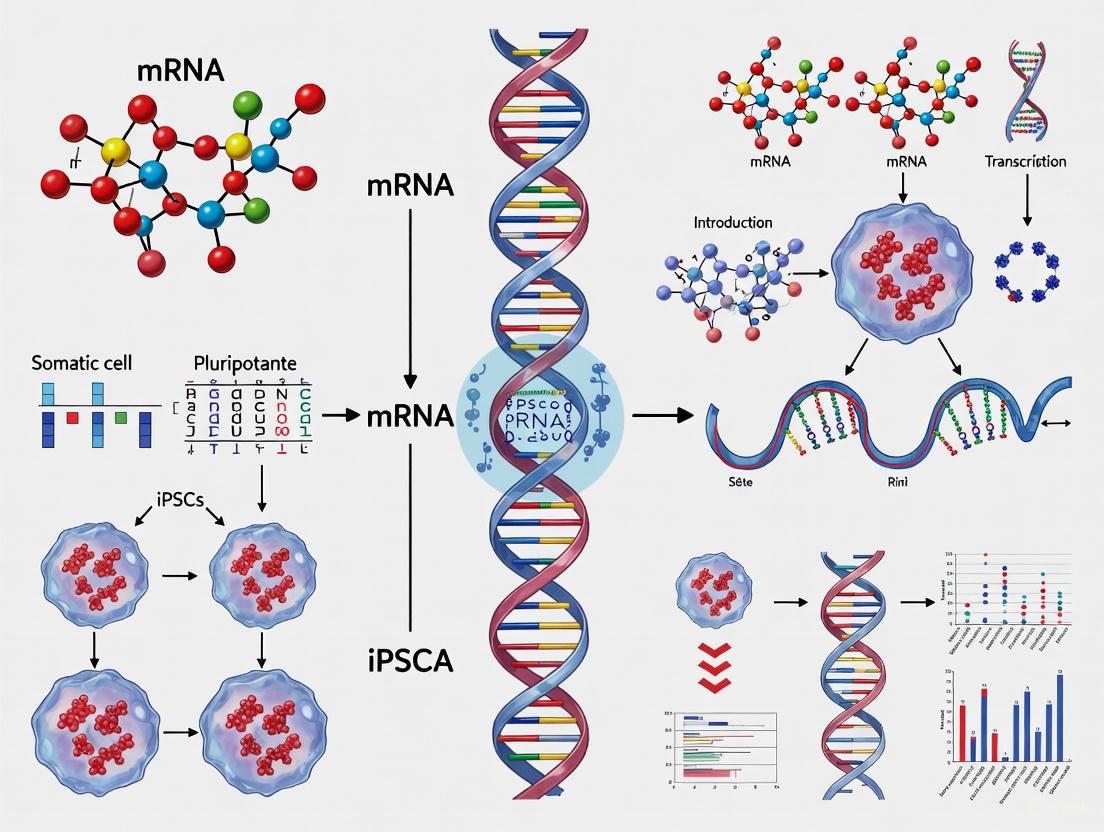
This article provides a comprehensive analysis of the transformative role of messenger RNA (mRNA) technology in the generation and application of induced pluripotent stem cells (iPSCs).
mRNA Reprogramming in iPSC Generation: Mechanisms, Clinical Applications, and Future Directions
Abstract
This article provides a comprehensive analysis of the transformative role of messenger RNA (mRNA) technology in the generation and application of induced pluripotent stem cells (iPSCs). Tailored for researchers, scientists, and drug development professionals, it explores the foundational science behind mRNA reprogramming, detailing its superiority as a non-integrating, footprint-free method for producing clinical-grade pluripotent stem cells. The scope encompasses current methodological protocols, key applications in disease modeling and regenerative medicine, and strategic solutions for overcoming technical challenges such as immunogenicity and low efficiency. Furthermore, the article presents a comparative evaluation of mRNA against other reprogramming systems, validating its position as a leading platform for developing safe, scalable, and personalized cell therapies.
From Yamanaka Factors to mRNA: Redefining the Foundations of Cellular Reprogramming
The discovery of induced pluripotent stem cells (iPSCs) by Shinya Yamanaka in 2006 represents a transformative breakthrough in regenerative medicine and cellular biology. This pioneering work demonstrated that somatic cells could be reprogrammed to an embryonic-like state through the introduction of just four transcription factors—Oct3/4, Sox2, Klf4, and c-Myc (collectively known as the Yamanaka factors or OSKM) [1] [2]. This revolutionary finding earned Yamanaka the Nobel Prize in Physiology or Medicine in 2012 and established a new paradigm for understanding cellular plasticity [1] [3].
The fundamental principle underlying iPSC technology is the reversal of developmental commitment, essentially "rewinding" the epigenetic clock of differentiated cells back to a pluripotent state [4] [3]. This process bypasses the ethical concerns associated with embryonic stem cells (ESCs) while providing a virtually unlimited source of patient-specific cells for research and therapeutic applications [1]. The original iPSC derivation method involved introducing the four Yamanaka factors into mouse embryonic fibroblasts using retroviral vectors, resulting in cells exhibiting ESC-like morphology, gene expression patterns, and developmental potential [1] [2].
Since their initial discovery, iPSCs have become indispensable tools for disease modeling, drug screening, and regenerative medicine [3] [5]. However, the original reprogramming methods raised significant safety concerns, particularly regarding the use of integrating viral vectors and the inclusion of oncogenes like c-Myc, which limited their clinical applicability [1] [4]. This review examines the molecular mechanisms of iPSC induction, analyzes the safety limitations of early reprogramming methods, and explores the development of safer approaches—with particular emphasis on mRNA-based technologies that align with the broader thesis of mRNA's expanding role in iPSC research.
Molecular Mechanisms of Cellular Reprogramming
The Yamanaka Factors and Their Functions
The four Yamanaka factors function as master regulators of pluripotency, each playing distinct yet complementary roles in the reprogramming process. Understanding their individual functions and synergistic interactions provides crucial insights into the molecular basis of cellular reprogramming.
Table 1: Core Pluripotency Transcription Factors and Their Functions
| Transcription Factor | Gene Family | Primary Function in Reprogramming | Notes on Requirement |
|---|---|---|---|
| Oct3/4 (Pou5f1) | POU-domain | Master regulator of pluripotency; essential for establishment and maintenance of pluripotent state | Absolutely required; absence leads to trophoblast differentiation [1] |
| Sox2 | SRY-related HMG-box | Partners with Oct3/4 to activate pluripotency network; maintains self-renewal | Critical core factor; other Sox family members (Sox1, Sox3, Sox15) can substitute with varying efficiency [1] |
| Klf4 | Krüppel-like factor | Promotes mesenchymal-to-epithelial transition (MET); modulates chromatin accessibility | Functionally replaceable by related factors Klf2, Klf5; can be omitted in certain cell types with endogenous expression [1] [4] |
| c-Myc | Myc proto-oncogene | Regulates metabolic reprogramming; promotes chromatin remodeling; enhances proliferation | Not absolutely required but significantly improves efficiency; concerning due to oncogenic potential [1] [4] [6] |
The reprogramming process occurs through a series of molecular events that can be broadly divided into early and late phases [3]. The early phase is characterized by the silencing of somatic genes and initial activation of early pluripotency-associated genes, a process that occurs somewhat stochastically due to inefficient access to closed chromatin regions by the exogenous transcription factors [3]. The late phase is more deterministic and hierarchical, involving establishment of a stable pluripotent state with activation of late pluripotency genes and reorganization of chromatin architecture [3]. Throughout this process, the Yamanaka factors collectively regulate a developmental signaling network composed of at least 16 crucial signaling pathways that maintain pluripotency in embryonic stem cells [6].
Alternative Factor Combinations
While the original OSKM combination remains the most widely used reprogramming cocktail, several alternative factor combinations have been identified. Most notably, James Thomson's group demonstrated that human fibroblasts could be reprogrammed using a different set of factors: Oct4, Sox2, Nanog, and Lin28 (OSNL) [1] [3]. The specific requirements for exogenous factors can also vary depending on the somatic cell type being reprogrammed, as some cells endogenously express one or more of these factors at sufficient levels [4]. For instance, fetal neural stem cells, which express high endogenous levels of Sox2, can be reprogrammed with Oct4 alone [4].
Figure 1: The Two-Phase Process of Somatic Cell Reprogramming to iPSCs. The early phase is stochastic and inefficient, while the late phase is more deterministic and hierarchical, culminating in established pluripotency.
Safety Concerns with Initial Reprogramming Methods
Limitations of Viral Vector Systems
The original iPSC generation method utilized retroviral vectors for factor delivery, which posed significant safety concerns for clinical translation. These vectors integrate randomly into the host genome, potentially disrupting tumor suppressor genes or activating oncogenes through insertional mutagenesis [4]. Although the transgenes are often epigenetically silenced once pluripotency is established, reactivation of reprogramming factors—particularly the oncogene c-Myc—upon differentiation raises serious concerns about tumorigenic potential [1] [4]. In fact, approximately 25% of mice transplanted with c-Myc-induced iPSCs developed lethal teratomas, highlighting this significant safety risk [1].
The efficiency of the original reprogramming process was remarkably low (0.01-0.1%), requiring prolonged culture periods that increased the likelihood of accumulating genetic and epigenetic abnormalities [1] [4]. This low efficiency was partially attributed to the activation of tumor suppressor pathways, particularly p53, in response to the forced expression of oncogenic factors like c-Myc and Klf4 [4]. Consequently, researchers often employed p53 knockdown to improve reprogramming efficiency, further compounding safety concerns by disabling a critical cellular defense mechanism against malignant transformation [4].
Addressing the Tumorigenicity Risk
The risk of tumor formation represents the most significant barrier to clinical application of iPSC-based therapies. This risk manifests through multiple mechanisms. First, residual undifferentiated iPSCs contaminating differentiated cell populations could proliferate uncontrollably and form teratomas after transplantation [7]. Second, the reprogramming process itself can introduce unintentional genetic changes that predispose cells to malignant transformation [7]. Third, the continued presence or reactivation of reprogramming factors—particularly the proto-oncogene c-Myc—can drive uncontrolled proliferation [1] [7].
Table 2: Safety Concerns with Initial iPSC Generation Methods
| Safety Concern | Underlying Cause | Potential Consequence |
|---|---|---|
| Insertional Mutagenesis | Random integration of viral vectors into host genome | Disruption of tumor suppressor genes or activation of oncogenes [4] |
| Oncogene Reactivation | Reactivation of c-Myc after differentiation | Uncontrolled cell proliferation and tumor formation [1] |
| Incomplete Reprogramming | Partial epigenetic remodeling | Unpredictable differentiation potential and functional abnormalities [4] |
| Genetic Instability | Extended in vitro culture periods | Accumulation of mutations that predispose to malignant transformation [7] |
| Teratoma Formation | Residual undifferentiated iPSCs in transplant populations | Benign or malignant tumor development after transplantation [7] |
Advancements in Safer Reprogramming Methodologies
Non-Integrating Vector Systems
To address the safety limitations of integrating viral vectors, researchers have developed numerous non-integrating delivery systems. These approaches can be broadly categorized into DNA-based, RNA-based, and protein-based methods, each with distinct advantages and limitations [4].
Episomal vectors are DNA-based systems that remain separate from the host genome and are gradually lost during cell divisions, eliminating the risk of insertional mutagenesis [4]. Sendai virus, an RNA virus that replicates in the cytoplasm without nuclear integration, has emerged as a particularly efficient system for generating footprint-free iPSCs [8]. The non-integrating lentiviral system represents another improvement, as these vectors are engineered to minimize integration while maintaining high transduction efficiency [4]. Additionally, excisable systems utilize loxP or frt sites to flank the reprogramming cassette in integrating vectors, allowing for its subsequent removal using Cre or Flp recombinase after reprogramming is complete [4].
The Emergence of mRNA-Based Reprogramming
Among the various non-integrating approaches, mRNA-based technology has emerged as a particularly promising strategy for safe and efficient cellular reprogramming. This method involves introducing in vitro transcribed mRNA molecules encoding the Yamanaka factors into somatic cells [8] [9]. Unlike DNA-based approaches, mRNA molecules do not require nuclear entry and are transiently expressed in the cytoplasm, eliminating the risk of genomic integration entirely [8] [9].
The mRNA reprogramming approach offers several distinct advantages. It demonstrates significantly faster reprogramming kinetics compared to other methods, with iPSC colonies typically appearing within two weeks [8]. The transient nature of mRNA expression allows for precise temporal control over factor expression, enabling researchers to optimize the duration and timing of reprogramming factor activity [9]. Perhaps most importantly, mRNA reprogramming completely eliminates the risk of insertional mutagenesis and does not leave a genetic "footprint" in the resulting iPSCs, addressing a critical safety concern for clinical applications [8] [9].
However, mRNA-based reprogramming also presents technical challenges. The foreign mRNA molecules can trigger innate immune responses in host cells, potentially reducing efficiency and viability [9]. This limitation has been addressed through modifications to mRNA chemistry, particularly the incorporation of modified nucleosides such as pseudouridine, which dampen immune recognition while enhancing translational efficiency [9]. Additionally, mRNA molecules have limited stability within cells, requiring repeated transfections to maintain sufficient reprogramming factor expression throughout the process [8] [9].
Figure 2: Evolution of iPSC Reprogramming Methods from Integrating Viral Vectors to Safer Non-Integrating Approaches, Including mRNA-Based Technology.
The Scientist's Toolkit: Essential Reagents for iPSC Generation
The successful generation and maintenance of iPSCs requires a carefully selected set of research reagents and tools. The following table summarizes key solutions and materials essential for iPSC work, particularly focusing on mRNA-based reprogramming approaches.
Table 3: Essential Research Reagents for mRNA-Based iPSC Generation
| Research Tool | Function | Application Notes |
|---|---|---|
| Modified mRNA | Encodes reprogramming factors (OCT4, SOX2, KLF4, c-MYC) without genomic integration | Nucleoside modifications (e.g., pseudouridine) reduce immunogenicity and enhance stability [8] [9] |
| Transfection Reagent | Facilitates cellular uptake of mRNA molecules | Must balance efficiency with cytotoxicity; multiple transfections typically required [8] |
| Immune Suppressors | Minimizes immune response to foreign mRNA | Small molecules (e.g., B18R) can inhibit antiviral pathways and improve cell viability [9] |
| Pluripotency Media | Supports growth and maintenance of emerging iPSCs | Typically contain bFGF and TGF-β; essential for establishing pluripotent state [1] [8] |
| Extracellular Matrix | Provides substrate for iPSC attachment and growth | Matrigel, vitronectin, or laminin-521 create optimal surface for pluripotent cells [8] |
| Pluripotency Markers | Validates successful reprogramming | Antibodies for OCT4, SOX2, NANOG, SSEA-4, TRA-1-60; essential for quality control [1] [3] |
| GMP-Compliant Materials | Ensures clinical-grade quality for therapeutic applications | Required for translation to clinical use; includes xeno-free reagents [8] [7] |
Current Applications and Future Directions
The development of safer reprogramming methods, particularly mRNA-based approaches, has accelerated the clinical translation of iPSC technology. Early clinical trials using iPSC-derived cells have emerged for conditions including heart failure, steroid-resistant graft-versus-host disease (GVHD), degenerative eye diseases, Parkinson's disease, and diabetes [7] [5]. Notably, the first formal clinical trial of an allogeneic iPSC-derived cell product (CYP-001 by Cynata Therapeutics) for treating GVHD met its clinical endpoints and produced positive safety and efficacy data, paving the way for advanced Phase 2 and 3 trials [5].
Beyond cellular therapies, iPSCs have become indispensable tools for disease modeling, drug screening, and toxicology testing [3] [5]. The combination of iPSC technology with CRISPR-Cas9 gene editing has been particularly transformative, enabling precise genetic modifications in patient-specific cells for both disease modeling and therapeutic correction [8]. Emerging applications include the generation of 3D organoids that recapitulate tissue architecture and function, creation of humanized disease models for drug screening, and even bioprinting of functional tissues [8] [5].
Future advancements in iPSC technology will likely focus on further improving the safety profile, standardization, and scalability of iPSC generation and differentiation. The integration of machine learning for quality control and optimization of differentiation protocols represents a promising direction for addressing current challenges in reproducibility and characterization [8]. Additionally, ongoing efforts to engineer hypoimmunogenic iPSCs through genetic modification of HLA genes may enable the creation of universal donor cell lines that evade immune rejection [8].
The iPSC field has undergone remarkable evolution since the initial discovery of the Yamanaka factors, transitioning from proof-of-concept experiments to sophisticated clinical applications. While the original reprogramming methods established the fundamental principles of cellular reprogramming, their safety limitations necessitated the development of more refined approaches. mRNA-based technology has emerged as a particularly promising strategy, offering a non-integrating, footprint-free method for generating clinical-grade iPSCs. As research continues to refine these techniques and address remaining challenges in tumorigenicity and standardization, iPSC-based approaches are poised to revolutionize regenerative medicine, disease modeling, and drug development, ultimately fulfilling the immense therapeutic potential first envisioned with Yamanaka's groundbreaking discovery.
Why mRNA? The Rationale for a Transient, Non-Integrating Reprogramming Vector
The discovery that ordinary somatic cells could be reprogrammed into induced pluripotent stem cells (iPSCs) represented a paradigm shift in regenerative medicine [10]. Shinya Yamanaka's groundbreaking work demonstrated that forced expression of a defined set of transcription factors (Oct4, Sox2, Klf4, and c-Myc, collectively known as the OSKM factors) could revert specialized cells back to an embryonic-like state [10] [11]. This breakthrough promised a future of patient-specific regenerative therapies based on immunologically compatible material. However, a significant barrier initially prevented clinical translation: the need for integrating viral vectors to deliver reprogramming factors, which entailed potentially oncogenic alteration of the host genome [10].
The scientific community has since pursued various "footprint-free" reprogramming strategies to overcome this limitation, including non-integrating DNA vectors, protein transduction, and RNA-based systems [10] [12]. Among these techniques, mRNA-based reprogramming has emerged as "the most unambiguously 'footprint-free,' most productive, and perhaps the best suited to clinical production of stem cells" [10]. This technical guide examines the scientific rationale for selecting mRNA as a transient, non-integrating vector for cellular reprogramming, detailing the mechanisms, methodologies, and advantages that position this technology as a cornerstone of modern iPSC research and therapeutic development.
The Integration Problem: Limitations of Conventional Reprogramming Vectors
Risks of Genome-Modifying Methods
Initial iPSC generation relied heavily on integrating retroviral and lentiviral vectors to achieve sustained expression of the Yamanaka factors [10] [12]. While these vectors provided the necessary persistent transgene expression required for reprogramming (a process taking 10-30 days), they presented substantial clinical risks:
- Insertional mutagenesis: Random integration of viral cassettes into the host genome can disrupt tumor suppressor genes or activate oncogenes [12]
- Uncontrolled transgene reactivation: Sporadic reactivation of silenced reprogramming factors, particularly the oncogene c-Myc, could lead to tumor formation in differentiated progeny [10]
- Unsilencing variability: The timing and efficiency of viral silencing is unpredictable, potentially inhibiting complete reprogramming or subsequent differentiation [12]
Even with advanced excisable systems (Cre-lox, PiggyBac), the need for additional manipulation and rigorous screening for complete excision complicates manufacturing and increases costs for clinical applications [12].
Limitations of Alternative Non-Integrating Methods
Several alternative approaches have been developed to address genomic integration concerns, though each presents distinct limitations:
Table 1: Comparison of Non-Integrating Reprogramming Methods
| Method | Key Features | Efficiency | Limitations |
|---|---|---|---|
| Adenoviral Vectors | Non-integrating DNA virus | Low (≤0.0001%) | Inconstant gene expression in dividing cells [10] [12] |
| Episomal Plasmids | DNA vectors with replication origin | 0.04-0.3% | Low efficiency; potential genomic recombination [10] [13] |
| Sendai Virus | RNA-based cytoplasmic virus | 0.01-1% | Difficult to clear persistent virus [10] [13] |
| Protein Transduction | Modified reprogramming proteins | Very low | Extremely low efficiency; technical complexity [10] |
| mRNA Reprogramming | Synthetic modified mRNA | High (1-4%) | Requires daily transfections; optimized reagent needed [10] [13] |
As evidenced in Table 1, mRNA reprogramming offers superior efficiency while maintaining a truly transient presence without genomic integration.
The mRNA Solution: Engineering a Transient Reprogramming Vector
Structural Engineering of Modified mRNA
Natural mRNA molecules present challenges for reprogramming applications, including instability, high immunogenicity, and difficulty in delivery [13]. Modern mRNA reprogramming protocols utilize synthetically modified mRNA (modRNA) that incorporates specific structural optimizations:
Figure 1: Engineered structure of modified mRNA for reprogramming. Key modifications enhance stability, translation efficiency, and reduce immunogenicity.
These structural modifications address the limitations of conventional mRNA:
- 5' Cap Modifications: Anti-reverse cap analogs (ARCAs) prevent reverse incorporation during transcription, ensuring proper recognition by translation initiation factors and protection from decapping enzymes [13]
- Nucleotide Substitution: Incorporation of modified nucleotides (5-methylcytidine [5mC], pseudouridine [ψU], N1-methylpseudouridine [1mψU]) reduces recognition by Toll-like receptors (TLR7, TLR8), minimizing innate immune activation [13]
- UTR Optimization: Implementation of highly stable UTR sequences derived from α- and β-globin genes enhances translational efficiency and mRNA half-life [13]
- Poly-A Tail Engineering: Tail lengths of 120-150 nucleotides protect against exonucleases and facilitate nuclear export [13]
Mechanism of Transient Reprogramming
The fundamental advantage of mRNA reprogramming lies in its purely cytoplasmic activity and transient nature. Unlike DNA-based methods, mRNA does not enter the nucleus and contains no elements capable of genomic integration [10] [13]. The mechanism follows this sequence:
- Cellular Uptake: Synthetic mRNA molecules are delivered via lipofection or electroporation
- Cytoplasmic Translation: Host ribosomes translate mRNA into functional reprogramming factor proteins
- Nuclear Translocation: Synthesized transcription factors migrate to the nucleus to initiate reprogramming
- mRNA Degradation: Natural mRNA decay mechanisms clear the vector within hours to days
- Sustained Expression: Repeated transfections maintain factor expression until endogenous pluripotency networks activate
This transient delivery provides precise control over reprogramming factor dosing, stoichiometry, and time course—critical parameters for efficient iPSC generation [10].
Experimental Implementation: mRNA Reprogramming Methodology
Core Reprogramming Workflow
Standard mRNA reprogramming protocols follow a systematic workflow to achieve efficient conversion of somatic cells to iPSCs:
Figure 2: mRNA reprogramming workflow. The process involves daily transfections until endogenous pluripotency factors maintain the reprogrammed state.
Essential Research Reagents and Materials
Successful implementation of mRNA reprogramming requires specific reagents optimized for this application:
Table 2: Essential Research Reagents for mRNA Reprogramming
| Reagent Category | Specific Examples | Function & Importance |
|---|---|---|
| Reprogramming mRNAs | Modified mRNA encoding OCT4, SOX2, KLF4, c-MYC, LIN28, NANOG [14] [13] | Core transcription factors for pluripotency induction; modified nucleotides reduce immunogenicity |
| Transfection Reagent | Lipid-based transfection reagents [10] | Enables efficient cellular uptake of mRNA molecules through formation of lipid nanoparticles |
| Cell Culture Medium | Pluripotent stem cell-specific medium [11] | Supports emergence and growth of iPSC colonies with appropriate nutrients and signaling factors |
| Supplemental Factors | B18R interferon inhibitor [13] | Counteracts residual innate immune response to enhance mRNA translation and cell viability |
| Basal Medium | DMEM/F12 or equivalent [11] | Serves as base for specialized reprogramming media formulations |
| Extracellular Matrix | Vitronectin, Matrigel, or Laminin-521 [11] | Provides substrate for cell attachment and signaling cues that support pluripotency |
| Small Molecules | VPA, 8-Br-cAMP, RepSox [11] | Epigenetic modifiers and signaling pathway inhibitors that enhance reprogramming efficiency |
Key Protocol Variations and Optimizations
Factor Cocktail Composition
While the original Yamanaka factors (OSKM) form the foundation, enhanced mRNA reprogramming protocols often include additional factors:
- OSKMMLN cocktail: OCT4, SOX2, KLF4, c-MYC, plus LIN28 and NANOG for improved human cell reprogramming efficiency [14]
- c-MYC variants: Substitution with L-MYC to reduce potential tumorigenicity while maintaining efficiency [11]
- Supplemental factors: Addition of GLIS1, ESRRB, or nuclear receptor NR5A2 to enhance reprogramming in specific cell types [11]
Transfection Schedule and Duration
The transient nature of mRNA necessitates precise timing:
- Daily transfections for 12-20 days, depending on cell type and efficiency [14]
- "Point of No Return" (PNR): Occurs approximately at day 5-7 when endogenous pluripotency factors become self-sustaining [14]
- Short-term application: Some applications use brief exposure (4 days) for partial reprogramming to reverse aging hallmarks without complete lineage conversion [14]
Immune Response Management
Despite nucleotide modifications, residual immune activation may occur:
- Interferon suppression: Addition of B18R protein (a type I interferon inhibitor) to culture medium [13]
- Dose optimization: Titration of mRNA concentrations to balance expression efficiency against immune activation
- Staggered introduction: Sequential rather than simultaneous introduction of factors to reduce cellular stress
Applications and Evidence: Validation of the mRNA Approach
Quantitative Assessment of Reprogramming Efficiency
Comparative studies demonstrate the advantages of mRNA reprogramming across multiple metrics:
Table 3: Performance Metrics of mRNA vs. Alternative Reprogramming Methods
| Performance Metric | mRNA Method | Integrating Viral Methods | Sendai Virus | Episomal Plasmid |
|---|---|---|---|---|
| Reprogramming Efficiency | 1-4% [10] [13] | 0.01-0.1% [13] | 0.01-1% [13] | 0.04-0.3% [13] |
| Time to iPSC Emergence | 12-20 days [14] | 20-30 days [10] | 18-25 days | 25-30 days |
| Genomic Integration Risk | None [10] [13] | High [10] [12] | None (but viral persistence) [13] | Low (but possible recombination) [10] |
| Footprint-Free Status | Complete [10] | None (without excision) [12] | Requires viral clearance [13] | Requires plasmid loss verification [10] |
| Tumorigenicity Risk | Very low [10] | High (transgene reactivation) [10] | Low | Low |
Functional Validation in Research and Applications
mRNA-derived iPSCs have been rigorously validated through multiple applications:
- Disease modeling: Patient-specific iPSCs for conditions including amyotrophic lateral sclerosis (ALS), with differentiation into disease-relevant cells like motor neurons [11]
- Aging reversal studies: Transient mRNA expression (4 days) reversed age-associated markers in human cells, including resetting the epigenetic clock by approximately 3-5 years [14]
- Clinical translation: Advancement to clinical trials for conditions including age-related macular degeneration, with the first transplant of iPSC-derived cells occurring in 2013 [5]
- Drug screening: Industrial-scale production of iPSCs and their differentiated progeny for compound screening and toxicology assessment [5] [15]
mRNA reprogramming represents the convergence of safety, efficiency, and precision in iPSC generation. The transient, non-integrating nature of the mRNA vector directly addresses the fundamental clinical safety concerns associated with earlier genome-modifying methods, while its high efficiency and controllability make it practically advantageous for both research and therapeutic applications.
As the field progresses toward clinical implementation, mRNA technology provides a versatile platform compatible with current Good Manufacturing Practice (GMP) standards and scalable production requirements [5] [15]. The ongoing refinement of mRNA design, delivery methods, and reprogramming protocols continues to enhance this approach, solidifying its role as a cornerstone methodology in the expanding landscape of iPSC-based research and regenerative medicine.
The demonstrated ability to precisely control reprogramming factor expression through mRNA delivery enables not only complete lineage conversion to pluripotency but also innovative applications in partial reprogramming and cellular rejuvenation [14]. This versatility, combined with the unequivocally transient nature of the vector, positions mRNA reprogramming as a fundamentally enabling technology for the next generation of iPSC applications in disease modeling, drug discovery, and cellular therapeutics.
The discovery that somatic cells can be reprogrammed into induced pluripotent stem cells (iPSCs) has revolutionized regenerative medicine and disease modeling. Among various reprogramming methods, synthetic modified mRNA technology represents a groundbreaking advance due to its high efficiency, non-integrating nature, and elimination of tumorigenic risks associated with viral vectors. This technical review examines the core mechanism by which synthetic mRNA drives cellular reprogramming, detailing the molecular events that enable the transition from a differentiated to pluripotent state. We explore how modified nucleobases evade innate immune recognition, facilitate sustained transgene expression, and ultimately activate the endogenous pluripotency network. The comprehensive analysis includes optimized experimental protocols, key signaling pathways, and quantitative data comparisons to provide researchers with a practical framework for implementing this transformative technology.
The generation of induced pluripotent stem cells (iPSCs) through the introduction of specific transcription factors represents one of the most significant breakthroughs in modern regenerative medicine. Since Shinya Yamanaka's initial demonstration that somatic cells could be reprogrammed using the OSKM factors (OCT4, SOX2, KLF4, and c-Myc), researchers have sought increasingly efficient and safe delivery methods [11] [1]. Traditional viral approaches have been hampered by several limitations: (1) permanent genomic integration with associated mutagenesis risks; (2) poor reprogramming efficiencies (typically 0.01%-0.1%); and (3) potential reactivation of oncogenes like c-Myc [16] [17].
Synthetic modified mRNA technology has emerged as a superior alternative that effectively addresses these limitations. This approach involves the in vitro transcription of mRNA sequences encoding reprogramming factors, with specific nucleobase modifications that prevent recognition by cellular innate immune sensors [16] [17]. The resulting "mod-RNA" can be transfected into target cells where it directs transient but highly efficient protein translation without any risk of genomic integration [18]. Initial studies demonstrated that this method could reprogram human fibroblasts with efficiencies dramatically surpassing established protocols—achieving conversion rates of 1-4% compared to 0.001-0.01% with viral methods [17]. Subsequent optimizations have pushed efficiencies even higher, with some protocols reporting successful reprogramming of up to 90.7% of individually plated primary fibroblasts [18].
The clinical implications of this technology are profound. RNA-induced pluripotent stem cells (RiPS cells) maintain genomic integrity and demonstrate a closer molecular resemblance to human embryonic stem cells than viral-derived iPSCs [17]. Furthermore, the same mRNA platform can subsequently direct the differentiation of RiPS cells into terminally differentiated cell types for therapeutic applications, creating a seamless, safe, and efficient pipeline for regenerative medicine [16].
Molecular Mechanisms of mRNA-Mediated Reprogramming
Overcoming Innate Immune Recognition
A fundamental challenge in exogenous mRNA delivery is the robust innate immune response that mammalian cells mount against foreign RNA. Conventional mRNA transfection typically triggers pattern recognition receptors (especially TLR and RIG-I receptors), leading to interferon secretion and eventual cell death [17]. The key innovation enabling mRNA-based reprogramming involves strategic nucleobase modifications, particularly replacing uridine with pseudouridine or similar analogs [16] [18]. These modified nucleobases effectively shield the mRNA from immune detection while enhancing translational capacity and RNA stability [17].
The mechanism operates through altered molecular structure that prevents binding to innate immune receptors while maintaining the coding capacity for protein translation. When Rossi and colleagues developed the first effective mRNA reprogramming system, they patented this modification technology specifically to avoid activating antiviral response pathways [17]. Without this critical modification, repeated transfections necessary for reprogramming would be impossible due to progressive cell death. The modified mRNA enables efficient protein expression for days and weeks without eliciting adverse cellular reactions, creating a permissive environment for the sustained expression of reprogramming factors needed to reshape cell identity [17].
Transcriptional Dynamics and Epigenetic Remodeling
The reprogramming process initiated by synthetic mRNA follows a biphasic trajectory with distinct molecular events:
Early Phase (Stochastic): During the initial days of transfection, the translated OSKM factors bind to partially accessible genomic loci, initiating suppression of somatic genes and early activation of pluripotency-associated genes. This phase is characterized by mesenchymal-to-epithelial transition (MET) and is highly dependent on factor stoichiometry [3]. The transient nature of mRNA-derived proteins creates a dynamic expression pattern that may actually enhance chromatin sampling by reprogramming factors.
Late Phase (Deterministic): After approximately 1-2 weeks, the process transitions to a more predictable phase where endogenous pluripotency factors become self-sustaining. Critical events include activation of the core pluripotency network (OCT4, SOX2, NANOG) and epigenetic remodeling that establishes stable pluripotency [19] [3]. The exogenous mRNA transfections can be discontinued once this autonomous pluripotent state is achieved.
Throughout both phases, the modified mRNA-encoded factors mediate extensive chromatin reconfiguration, including histone modification changes and DNA demethylation at pluripotency promoter regions [3]. The high efficiency of mRNA reprogramming is attributed to the robust, sustained protein expression that drives these epigenetic changes more rapidly than traditional methods.
Table 1: Key Molecular Events in mRNA-Mediated Reprogramming
| Reprogramming Phase | Timeframe | Major Molecular Events | Critical Factors |
|---|---|---|---|
| Early/Stochastic | Days 1-7 | Initiation of somatic gene silencing; Beginning of MET; Early pluripotency gene activation | OCT4, SOX2 with KLF4 |
| Transition | Days 7-14 | Establishment of epithelial phenotype; Enhanced chromatin accessibility; Activation of endogenous pluripotency factors | c-MYC (enhances chromatin opening) |
| Late/Deterministic | Days 14-21 | Stable endogenous pluripotency network; Epigenetic remodeling; Colony formation | Endogenous OCT4, SOX2, NANOG |
| Maturation | Days 21-28 | Stabilization of pluripotent state; Metabolic reprogramming; Preparation for differentiation | Self-renewal transcription factors |
Synergistic Enhancement with microRNAs
Recent optimizations have demonstrated that combining modified mRNA with specific microRNA mimics can synergistically enhance reprogramming efficiency. The addition of miR-367/302 family mimics to the standard mRNA cocktail has been shown to increase iPSC generation dramatically [18]. This particular miRNA family promotes reprogramming through multiple mechanisms: (1) targeting epigenetic regulators like AOF1 to facilitate chromatin remodeling; (2) suppressing differentiation pathways; and (3) enhancing the expression of key pluripotency factors [18].
The combination approach has yielded remarkable efficiencies—producing up to 4,019 iPSC colonies from just 500 starting human primary neonatal fibroblasts, representing reprogramming of up to 90.7% of individually plated cells [18]. This synergistic effect underscores the multi-layered nature of cellular reprogramming, where coordinated regulation at transcriptional and post-transcriptional levels generates the most robust pluripotency induction.
Experimental Protocols and Workflows
Optimized mRNA Reprogramming Protocol
The following workflow represents the current state-of-the-art for synthetic mRNA-based reprogramming of human primary fibroblasts, incorporating critical optimizations from recent studies:
Critical Protocol Parameters:
- Cell Density: 500 cells per well of a 6-well plate (significantly lower than traditional methods) [18]
- Transfection Buffer: Opti-MEM adjusted to pH 8.2 (critical for efficiency) [18]
- RNA Cocktail: 600 ng 5fM3O mod-mRNA (OCT4 variant, SOX2, KLF4, cMYC, LIN28A, NANOG) + 20 pmol miRNA-367/302 mimics [18]
- Transfection Interval: Every 48 hours for 7 sessions [18]
- Culture Medium: Knock-out serum replacement (KOSR) based reprogramming medium [18]
This protocol has been specifically optimized for human primary fibroblasts, which historically proved more resistant to mRNA reprogramming than established fibroblast cell lines. The adjustment of transfection buffer pH to 8.2 was found to dramatically improve transfection efficiency without increasing cytotoxicity, while the lower seeding density allows for more cell divisions during the reprogramming process—a key factor in successful epigenetic remodeling [18].
Enhanced Workflow with microRNA Synergy
The synergistic protocol incorporating microRNA mimics represents the current gold standard for efficiency. The microRNA component enhances reprogramming through several distinct mechanisms:
- Accelerated epigenetic remodeling by targeting chromatin modifiers
- Suppression of alternative differentiation pathways
- Stabilization of pluripotency factor expression
- Enhanced cell cycle progression in emerging iPSCs
This combination approach achieves what individual methods cannot accomplish alone, creating a permissive environment where a majority of transfected cells successfully complete the reprogramming journey [18].
Quantitative Analysis of Reprogramming Efficiency
Table 2: Comparative Efficiency of iPSC Reprogramming Methods
| Reprogramming Method | Efficiency Range | Reprogramming Time | Genomic Integration | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| Retroviral Vectors | 0.01%-0.1% | 3-4 weeks | Yes | Established protocol; Effective factor delivery | Insertional mutagenesis; Transgene reactivation |
| Lentiviral Vectors | 0.01%-0.2% | 3-4 weeks | Yes | Can reprogram non-dividing cells; Stable integration | Insertional mutagenesis; Complex vector design |
| Sendai Virus | 0.1%-1% | 2-4 weeks | No | High efficiency; Broad cell tropism | Viral clearance required; Potential immunogenicity |
| Episomal Plasmids | 0.001%-0.1% | 4-5 weeks | No | Non-viral; Simple delivery | Low efficiency; Multiple transfections often needed |
| Synthetic Modified mRNA | 1%-4% (standard) Up to 90.7% (optimized) | 2-3 weeks | No | Highest efficiency; No genomic integration; Precise control | Requires multiple transfections; Optimization needed for different cell types |
The quantitative advantage of synthetic mRNA reprogramming is unmistakable when comparing these methodologies. While standard mRNA protocols already achieve a 10 to 100-fold improvement over viral methods, the optimized protocols with microRNA synergy reach unprecedented efficiencies—essentially converting the majority of starting cells into iPSCs [18]. This high efficiency makes the technology particularly valuable for working with precious patient samples where cell numbers may be limited.
The timing of iPSC emergence also differs significantly between methods. mRNA-reprogrammed cells typically appear within 2-3 weeks, compared to 3-5 weeks for many other non-integrating methods [16] [18]. This accelerated timeline likely reflects the rapid, high-level protein expression achievable with modified mRNA and the more direct activation of the pluripotency network without the epigenetic barriers posed by DNA-based methods.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Synthetic mRNA Reprogramming
| Reagent Category | Specific Product/Component | Function in Reprogramming | Optimization Notes |
|---|---|---|---|
| Reprogramming Factors | 5fM3O mod-mRNA cocktail (OCT4 variant, SOX2, KLF4, cMYC, LIN28, NANOG) | Core transcription factor delivery | M3O is OCT4 fused with MyoD transactivation domain for enhanced activity [18] |
| Enhancing miRNAs | miR-367/302 family mimics | Synergistic enhancement of reprogramming efficiency | 20 pmol per transfection optimal; enables single-cell reprogramming [18] |
| Transfection Reagent | Lipofectamine RNAiMAX | RNA delivery into target cells | Superior performance with pH-adjusted buffers [18] |
| Transfection Buffer | Opti-MEM (pH adjusted to 8.2) | Medium for RNA-lipid complex formation | pH adjustment critical for primary fibroblast transfection efficiency [18] |
| Culture Medium | KOSR-based reprogramming medium | Supports emerging iPSCs and reprogramming process | Formulation optimized for low-density plating [18] |
| Cell Culture Matrix | Matrigel or recombinant vitronectin | Extracellular matrix support for feeder-free culture | Essential for clinical-grade iPSC generation [18] |
Applications and Future Directions
The implementation of synthetic mRNA reprogramming extends across multiple research and therapeutic domains:
Disease Modeling and Drug Discovery
iPSCs generated via mRNA technology offer particular advantages for disease modeling and pharmaceutical applications. The absence of genomic integration means that resulting iPSCs have unmodified genetic backgrounds, crucial for accurate disease phenotyping [16] [3]. Neurological diseases like amyotrophic lateral sclerosis (ALS) have been successfully modeled using iPSC-derived motor neurons created through mRNA reprogramming, enabling mechanistic studies and drug screening [11]. The technology facilitates the creation of patient-specific iPSC banks that preserve the exact genetic makeup of donors without viral-induced mutations [19].
Clinical Translation and Regenerative Medicine
The path to clinical application requires methods that generate clinically compliant cell products. Synthetic mRNA reprogramming fulfills key safety criteria for regenerative medicine by eliminating integrating vectors and oncogenic transgene persistence [20] [8]. Clinical trials are already underway using iPSC-derived dopaminergic neurons for Parkinson's disease and retinal pigment epithelial cells for age-related macular degeneration [20]. The same mRNA technology used for reprogramming can subsequently direct the differentiation of iPSCs into therapeutic cell types, creating an integrated manufacturing platform [16] [17].
Emerging Technological Convergence
Future developments will likely combine mRNA reprogramming with other cutting-edge technologies. CRISPR-Cas9 gene editing can be performed on mRNA-generated iPSCs to correct genetic defects before differentiation and transplantation [20] [8]. Machine learning algorithms are being developed to automate the identification of optimal iPSC colonies and predict differentiation outcomes, potentially working in concert with mRNA-based protocols [20] [8]. The convergence of these technologies accelerates the movement toward personalized regenerative treatments tailored to individual patients.
Synthetic modified mRNA technology represents a paradigm shift in somatic cell reprogramming, offering an unprecedented combination of efficiency, safety, and precision. The core mechanism—bypassing innate immune recognition through nucleobase modifications while enabling transient but robust expression of reprogramming factors—addresses the fundamental limitations of previous approaches. The optimization of transfection parameters, combined with synergistic microRNA enhancement, now enables reliable reprogramming of even refractory primary human cells at remarkable efficiencies. As this technology continues to converge with advances in gene editing and artificial intelligence, it promises to accelerate both basic research and clinical applications of iPSC technology, ultimately fulfilling the promise of personalized regenerative medicine.
The clinical application of induced pluripotent stem cells (iPSCs) has been fundamentally constrained by the risk of tumorigenicity associated with genome-integrating vectors used in conventional reprogramming methods. This whitepaper delineates the key historical developments that established mRNA-based reprogramming as a premier "footprint-free" solution for generating clinically relevant iPSCs. We examine the molecular basis of this technology, provide a comparative analysis with other non-integrating methods, detail optimized experimental protocols, and present essential reagent solutions. The emergence of mRNA reprogramming represents a paradigm shift, enabling the production of iPSCs without genomic alterations and thereby accelerating the path toward personalized regenerative medicine.
The groundbreaking discovery by Takahashi and Yamanaka that somatic cells could be reprogrammed into induced pluripotent stem cells (iPSCs) by forced expression of defined transcription factors (Oct4, Sox2, Klf4, and c-Myc, collectively known as OSKM) heralded a new era in regenerative medicine [3]. This technology promised an unlimited supply of patient-specific cells for disease modeling, drug screening, and cellular therapies. However, the initial dependence on integrating retroviral and lentiviral vectors for factor delivery posed a significant clinical barrier due to the risk of insertional mutagenesis and potential tumorigenicity [10].
This safety concern catalyzed a dedicated research focus on achieving "footprint-free" reprogramming—methods that could revert somatic cells to pluripotency without leaving permanent genetic modifications. Among the various techniques developed, including episomal vectors, Sendai virus, and protein transduction, mRNA-based reprogramming has emerged as particularly promising due to its completely non-integrating nature, high efficiency, and controllable kinetics [10] [8]. This document traces the historical trajectory of this solution, its technical refinements, and its current standing in the field of iPSC research and development.
Historical Trajectory of Reprogramming Methods
The evolution of reprogramming methods reflects a concerted effort to balance efficiency with clinical safety, moving progressively from integrating vectors to transient delivery systems.
The Integrating Vector Era
Yamanaka's original 2006-2007 work utilized retroviruses to deliver the OSKM factors. These vectors integrate into the host genome, enabling sustained expression necessary for the weeks-long reprogramming process. While this method proved the principle of cellular reprogramming, the permanent alteration of the host genome presented an unacceptable clinical risk, as silencing of the transgenes is often incomplete, and random integration can disrupt tumor suppressor genes or activate oncogenes [10] [3].
The Emergence of Non-Integrating Approaches
The field rapidly pursued alternative strategies to overcome these limitations, leading to several "footprint-free" methodologies, each with distinct advantages and drawbacks, as summarized in Table 1.
Table 1: Comparison of Footprint-Free Reprogramming Methods
| Method | Mechanism | Relative Efficiency | Genomic Integration? | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| Plasmid/Adenovirus [10] | DNA vector maintained episomally | Low | Very low frequency | Simple production | Inconstant gene expression in dividing cells |
| Episomes [10] | DNA vector with eukaryotic origin of replication | Moderate | Low frequency | Improved yield over plasmids | Requires rigorous genomic screening |
| Sendai Virus [10] [8] | Cytoplasmic RNA virus | High | No | Single delivery, high efficiency | Replication-deficient but requires dilution/selection to clear |
| Protein Transduction [10] | Cell-penetrating peptides | Very Low | No | Highest safety profile | Extremely low efficiency, technically challenging |
| Synthetic mRNA [10] [8] | Direct delivery of modified mRNA | High | No | Unambiguous footprint-free nature, controllable dosing | Requires multiple transfections, requires suppression of innate immunity |
The first compelling "footprint-free" solutions involved plasmid vectors and adenoviruses, but these suffered from low efficiency and the lingering, albeit small, risk of genomic integration [10]. The subsequent development of mRNA reprogramming addressed these shortcomings by offering a system where the reprogramming vector is a synthetic, transient molecule that functions exclusively in the cytoplasm and is degraded by natural cellular processes, leaving no genetic trace [10].
The Molecular Basis of mRNA Reprogramming
The core principle of mRNA reprogramming is the direct delivery of in vitro transcribed (IVT) mRNA molecules encoding the Yamanaka factors into the cytoplasm of target somatic cells. The host cell's translation machinery then produces the transcription factor proteins, which migrate to the nucleus to initiate the complex process of epigenetic remodeling and gene expression changes that lead to pluripotency.
A critical breakthrough that made this approach feasible was the mitigation of the innate immune response. Unmodified exogenous mRNA is recognized by pattern recognition receptors in the cell, triggering a potent antiviral response, including the production of type I interferons, which leads to global translational shutdown and apoptosis [10] [21]. This response was overcome through two key innovations:
- Nucleoside Modification: Incorporating modified nucleosides, such as pseudouridine, into the IVT mRNA effectively evades cellular immune sensors, dramatically reducing interferon signaling and enhancing mRNA stability and translational capacity [21].
- Optimized mRNA Structure: The synthetic mRNA is engineered to mimic mature native mRNA, featuring a 5' cap structure (e.g., Cap1), optimized 5' and 3' untranslated regions (UTRs), a protein-encoding open reading frame (ORF), and a poly(A) tail. These elements work in concert to promote ribosome binding, protect from exonuclease degradation, and boost protein expression [21].
The following diagram illustrates the workflow and key molecular components of the mRNA reprogramming process.
Technical Refinements and Protocol Standardization
Since its initial demonstration, the mRNA reprogramming protocol has been refined to improve its efficiency, reproducibility, and ease of use. A significant advancement was the development of a single, self-replicating RNA vector based on the Venezuelan equine encephalitis virus, which enables sustained expression of the reprogramming factors from a single transfection, simplifying the process considerably [22].
Detailed Experimental Protocol
The following is a generalized protocol for footprint-free iPSC generation using synthetic modified mRNA.
Key Reagent Solutions:
- Synthetic Modified mRNA: Commercially available kits or custom-synthesized mRNAs for OCT4, SOX2, KLF4, c-MYC, and optionally, LIN28 and a GFP reporter. Nucleotides are modified (e.g., with pseudouridine) to reduce immunogenicity.
- Transfection Reagent: A reagent suitable for sensitive primary cells (e.g., lipid-based nanoparticles or other commercial transfection kits).
- Cell Culture Medium: Somatic cell medium (e.g., fibroblast growth medium), mRNA reprogramming medium (specialized medium supporting pluripotency), and iPSC maintenance medium (e.g., mTeSR or E8).
- Immune Suppression Supplement: A solution of interferon suppressor (e.g., B18R protein, which binds and inhibits type I interferons) is often added to the medium during the initial transfection phases.
Procedure:
- Cell Seeding: Seed human dermal fibroblasts (HDFs) or other target somatic cells at an appropriate density (e.g., 10,000-50,000 cells per well of a 24-well plate) in their standard growth medium. Incubate for 24 hours to allow cells to adhere and reach 50-70% confluence.
- Transfection Cycle: a. Prepare transfection complexes by mixing the cocktail of modified mRNAs (a total of 0.5-1 µg per well) with the transfection reagent in a serum-free medium, following the manufacturer's instructions. b. Replace the cell culture medium with fresh medium supplemented with the interferon suppressor (e.g., 100 ng/mL B18R). c. Add the transfection complexes dropwise to the cells. d. Incubate the cells for 24 hours.
- Medium Change: After 24 hours, replace the medium with fresh mRNA reprogramming medium containing the interferon suppressor.
- Repetition: Repeat the transfection cycle (Steps 2-3) daily for a period of 12-18 days. The daily transfections are critical to maintain a high intracellular concentration of the reprogramming proteins.
- Colony Observation and Picking: Between days 7 and 21, compact, embryonic stem cell-like colonies will emerge. Once colonies reach a sufficient size, pick them mechanically or enzymatically and transfer them to a feeder-free culture system (e.g., on Matrigel-coated plates) in iPSC maintenance medium.
- Characterization: Expand clonal lines and characterize them for pluripotency markers (e.g., immunocytochemistry for NANOG, TRA-1-60, SSEA4; RT-PCR for endogenous pluripotency genes) and functional capacity (e.g., in vitro differentiation into trilineage embryoid bodies).
Table 2: Key Research Reagent Solutions for mRNA Reprogramming
| Reagent Category | Specific Example | Function in Protocol |
|---|---|---|
| Reprogramming mRNAs | Modified mRNA for OCT4, SOX2, KLF4, c-MYC [10] [22] | Encodes the transcription factors required to induce pluripotency. Modified bases reduce innate immune recognition. |
| Transfection Vehicle | Lipid-Based Transfection Reagent [10] | Forms complexes with mRNA to facilitate its efficient delivery across the cell membrane. |
| Immune Suppressor | B18R Interferon Inhibitor [10] | A recombinant protein that binds to and neutralizes type I interferons, preventing translational shutdown and cell death. |
| Reprogramming Medium | Commercial Pluripotency Support Medium | A specialized medium formulation that supports the metabolic and signaling needs of reprogramming cells and emerging iPSCs. |
| Culture Substrate | Matrigel, Laminin-521 | Provides a defined, feeder-free extracellular matrix for the attachment and growth of iPSCs. |
The emergence of mRNA-based reprogramming represents a definitive solution to the critical challenge of genomic modification in iPSC generation. Its unambiguous "footprint-free" character, combined with high efficiency and controllable kinetics, makes it ideally suited for clinical-grade iPSC production [10] [8]. The technology has matured from a proof-of-concept to a robust, industrialized process, with ongoing efforts focused on further simplifying the protocol (e.g., via self-replicating RNAs) and optimizing it for specific clinical applications.
As the field of regenerative medicine advances toward clinical trials for conditions like age-related macular degeneration, Parkinson's disease, and ischemic heart disease, the availability of a safe, reliable, and non-integrating reprogramming method is paramount. mRNA technology not only fulfills this requirement but also exemplifies the power of synthetic biology to overcome fundamental biological barriers, solidifying its role as a cornerstone of modern iPSC research and therapeutic development.
Protocols and Pipelines: Implementing mRNA Reprogramming in Research and Therapy
The generation of induced pluripotent stem cells (iPSCs) represents one of the most significant breakthroughs in regenerative medicine, enabling the reprogramming of somatic cells into a pluripotent state. Within this field, messenger RNA (mRNA) transfection has emerged as a pivotal technology for clinical-grade iPSC generation due to its superior safety profile and high efficiency. Unlike early viral vector methods that posed risks of genomic integration and insertional mutagenesis, mRNA-based delivery offers a non-integrating approach that minimizes oncogenic risk while providing transient, high-level expression of reprogramming factors [11] [8]. This technical guide details a standardized mRNA transfection protocol optimized for generating clinical-grade iPSCs, framing this methodology within the broader thesis that mRNA technology is fundamentally advancing iPSC research by balancing efficiency with safety—a critical requirement for therapeutic applications.
The foundational principle of mRNA reprogramming involves introducing synthetic, modified mRNA molecules encoding key pluripotency factors into somatic cells. These mRNA molecules are translated into proteins that orchestrate the epigenetic and transcriptional remodeling necessary to revert differentiated cells to a pluripotent state. Through advancements in mRNA chemistry, delivery platforms, and manufacturing consistency, researchers can now achieve reprogramming efficiencies exceeding 10% while maintaining compliance with Good Manufacturing Practice (GMP) standards [8] [23]. This protocol leverages lipid nanoparticle (LNP) formulations to protect mRNA cargo and facilitate efficient cellular uptake, providing researchers with a robust workflow suitable for both autologous and allogeneic therapy development.
Core Principles of mRNA Reprogramming
Advantages Over Conventional Methods
mRNA-based reprogramming offers distinct advantages that make it particularly suitable for clinical-grade iPSC generation. The non-integrating nature of mRNA eliminates the risk of permanent genetic alterations in target cells, addressing a primary safety concern associated with viral vectors [8]. Furthermore, mRNA transfection provides precise temporal control over reprogramming factor expression, allowing researchers to optimize the duration and levels of protein expression to mimic natural developmental transitions [11]. The transient presence of exogenous mRNA—typically degraded within days—reduces the potential for incomplete reprogramming or aberrant persistence of reprogramming factors that might impede differentiation capacity [23].
From a manufacturing perspective, mRNA platforms offer superior scalability and reproducibility compared to viral systems. The production of synthetic mRNA is highly standardized, with well-established quality control parameters that ensure batch-to-batch consistency—a critical requirement for clinical translation [8] [23]. Additionally, mRNA-based approaches avoid the biosafety complications associated with viral production systems, simplifying regulatory approval pathways. These collective advantages position mRNA transfection as the leading technology for generating research-grade and clinical-grade iPSCs, particularly as the field advances toward broader therapeutic implementation.
Essential Reprogramming Factors
The core reprogramming process relies on the introduction of factors that reset epigenetic memory and activate pluripotency networks. While various factor combinations have been explored, the most established formulations are based on the original Yamanaka factors with modifications to enhance safety and efficiency:
Table 1: Key Reprogramming Factors and Their Functions
| Factor | Primary Function | Safety Considerations |
|---|---|---|
| OCT4 (POU5F1) | Master regulator of pluripotency; initiates epigenetic remodeling | Essential; no known alternatives |
| SOX2 | Partners with OCT4; activates pluripotency network | Critical; SOX1/SOX3 can partially substitute |
| KLF4 | Promotes proliferation; suppresses somatic gene expression | KLF2/KLF5 can substitute with reduced efficiency |
| c-MYC | Enhances proliferation; alters metabolism | Oncogenic potential; L-MYC or N-MYC are safer alternatives |
| LIN28 | Regulates miRNA processing; promotes cell cycle progression | Often used with NANOG in OSNL combination |
Recent advances have demonstrated that OCT4 alone may be sufficient for reprogramming certain cell types, particularly neural stem cells, highlighting its central role in the process [11]. However, for most somatic cell sources, combinations of four to five factors delivered via mRNA achieve optimal efficiency. The OSKML combination (OCT4, SOX2, KLF4, c-MYC/L-MYC, LIN28) has demonstrated particular effectiveness, especially for challenging cell types like those from aged donors or patients with underlying medical conditions [19]. The specific ratio of these factors is critically important, as studies have shown that the OCT4:SOX2 expression ratio significantly influences both reprogramming efficiency and the quality of resulting iPSC colonies [19].
The Scientist's Toolkit: Essential Reagents and Materials
The successful implementation of mRNA reprogramming requires carefully selected reagents and materials that maintain stringent quality standards, particularly for clinical-grade applications. The following toolkit comprises essential components for the protocol:
Table 2: Research Reagent Solutions for mRNA Reprogramming
| Reagent Category | Specific Examples | Function & Importance |
|---|---|---|
| Mammalian Cells | Human dermal fibroblasts, PBMCs | Starting somatic cell source; require validation and screening |
| Reprogramming mRNA Cocktail | uBriGene iPSC-RPM-mRNA-LNP-FB, iPSC-RPM-RNA-LNP-V2 | Core reprogramming factors in LNP delivery system |
| Culture Medium | ReproTeSR, StemMACS CardioDiff | Supports reprogramming and maintenance; xeno-free for clinical use |
| Culture Vessels | BioLaminin 521-coated plates, iMatrix-511 | ECM coating for feeder-free culture; defined composition |
| Enhancer Molecules | B18R protein, Serum-Free Enhancer B | Increases efficiency; counters innate immune response |
| Transfection System | Lipid nanoparticles (LNPs) | Protects mRNA and facilitates cellular uptake |
| Characterization Tools | Alkaline phosphatase staining, SSEA4/TRA-1-81 antibodies | Confirms pluripotency marker expression |
The selection of GMP-grade reagents throughout the workflow is essential for clinical translation. This includes using xeno-free culture components, validated sourcing of critical reagents like the mRNA-LNP cocktail, and implementing rigorous quality control testing for each component [23] [24]. Additionally, the protocol incorporates enhancer molecules such as B18R (a type I interferon inhibitor) that significantly improve viability and reprogramming efficiency by mitigating the innate immune response to exogenous mRNA [23].
mRNA Transfection Protocol: Detailed Step-by-Step Workflow
Pre-Programming Phase: Cell Preparation and Quality Control
Day -3 to Day -1: Cell Thawing and Expansion
- Begin with cryopreserved somatic cells (fibroblasts or PBMCs) from validated sources. For fibroblasts, thaw and plate at 15,000-20,000 cells/cm² on BioLaminin 521-coated plates in fibroblast culture medium. For PBMCs, use non-tissue culture treated plates with PBMC-specific medium containing appropriate cytokines [23].
- Culture cells for 2-3 passages to ensure recovery, proliferation competence, and absence of microbial contamination. Perform viability assessment (≥90% required) and check for morphological abnormalities.
- One day prior to reprogramming initiation, ensure cells are 70-80% confluent and in log-phase growth. Replace medium with richer alternative medium (e.g., supplemented with 8-Br-cAMP and valproic acid) to precondition cells and enhance reprogramming competence [11].
Quality Control Checkpoints:
- Verify mycoplasma-free status using standardized testing methods.
- Confirm cell identity through short tandem repeat (STR) profiling.
- Document population doubling times and morphological characteristics.
Programming Phase: mRNA Transfection and Colony Emergence
Day 0 to Day 8: Daily mRNA Transfection
- For fibroblasts: Aspirate medium and add fresh ReproTeSR medium supplemented with B18R (200ng/mL). Add mRNA-LNP cocktail directly to culture medium at optimized volume (e.g., 60μL per well of 24-well plate) [23]. Gently swirl plate to ensure even distribution.
- For PBMCs: Centrifuge cells at 300×g for 5 minutes, resuspend in ReproTeSR medium with Serum-Free Enhancer B, and add RNA-LNP cocktail every other day (total of four treatments) [23].
- Repeat transfection procedure daily for eight consecutive days (fibroblasts) or every other day (PBMCs). Maintain cells in humidified incubator at 37°C, 5% CO₂ between transfections.
- Monitor daily for morphological changes indicative of reprogramming initiation: increased nuclear-to-cytoplasmic ratio, emergence of small, compact cells with prominent nucleoli.
Day 9 to Day 15: Colony Formation and Maturation
- On day 9, switch to ReproTeSR medium without B18R. Continue daily medium changes.
- By approximately day 11, distinct iPSC-like colonies should emerge with defined borders and typical embryonic stem cell morphology.
- Around day 13-15, colonies should expand sufficiently for picking (typically 0.5-1mm diameter with high cell density).
The following workflow diagram illustrates the complete reprogramming process:
Post-Programming Phase: Colony Picking and Expansion
Day 16 to Day 20: Colony Selection and Expansion
- Prepare new BioLaminin 521-coated plates for colony transfer.
- Manually pick well-defined iPSC colonies using sterile pipette tips or specialized picking tools under microscopic visualization.
- Transfer individual colonies to separate wells of 48- or 24-well plates containing pre-warmed ReproTeSR or similar maintenance medium.
- Monitor attachment and expansion daily, with first passaging typically required 5-7 days after picking.
Quality Assessment and Banking
- Upon reaching 70-80% confluence in 6-well plates, confirm pluripotency through alkaline phosphatase staining and immunocytochemistry for markers including SSEA4, TRA-1-81, SOX2, and NANOG [23].
- Perform comprehensive characterization including karyotyping, STR analysis, and mycoplasma testing.
- For clinical applications, establish master and working cell banks using standardized cryopreservation protocols with defined, xeno-free cryoprotectants.
Troubleshooting and Optimization Strategies
Even with optimized protocols, researchers may encounter challenges that require specific interventions:
Table 3: Troubleshooting Common Issues in mRNA Reprogramming
| Problem | Potential Causes | Solutions |
|---|---|---|
| Low Transfection Efficiency | Poor mRNA quality, suboptimal LNP:cell ratio, impaired cellular uptake | Validate mRNA integrity, titrate LNP concentration, ensure cells are in log-phase growth |
| High Cell Death | Cytotoxicity of transfection reagent, innate immune activation | Include B18R in medium, optimize cell density, use serum-free Enhancer B for difficult cells |
| Incomplete Reprogramming | Insufficient factor expression, inadequate duration, suboptimal factor ratio | Extend transfection period, verify factor ratios, include additional factors (LIN28, NANOG) |
| Low Colony Numbers | Poor starting cell viability, inadequate culture conditions, donor-specific factors | Pre-condition with 8-Br-cAMP and VPA, use specialized kits for aged/diseased cells [11] [23] |
| Spontaneous Differentiation | Suboptimal colony picking timing, inappropriate culture conditions | Pick colonies earlier (day 16-18), ensure daily medium changes, use fresh matrix coating |
For challenging cell sources such as those from aged donors or patients with underlying diseases, the PBMC-specific kit (e.g., uBriGene iPSC-RPM-RNA-LNP-V2) with Enhancer B is recommended to overcome inherent reprogramming barriers [23]. Additionally, incorporating small molecules that modulate key signaling pathways—including Wnt, TGF-β, and BMP—can further enhance reprogramming efficiency and consistency across different cell sources [8].
The mRNA transfection protocol detailed herein represents a robust, standardized approach for generating clinical-grade iPSCs that aligns with the evolving regulatory landscape for cell-based therapies. By leveraging the precision and safety of mRNA technology, researchers can now produce iPSC lines with reduced oncogenic risk while maintaining high efficiency—addressing two critical challenges that have historically impeded clinical translation.
As the field advances, the integration of mRNA-based reprogramming with emerging technologies like CRISPR-Cas9 gene editing and 3D organoid culture will further expand the applications of iPSCs in disease modeling, drug screening, and regenerative medicine [8] [25]. The development of GMP-compliant mRNA reagents and standardized protocols ensures that research findings can be seamlessly translated to clinical applications, accelerating the realization of personalized regenerative therapies. Through continued refinement of mRNA design, delivery systems, and manufacturing processes, this technology platform is poised to remain central to iPSC research and its therapeutic implementations in the coming decade.
The discovery that somatic cells can be reprogrammed into induced pluripotent stem cells (iPSCs) using messenger RNA (mRNA) encoding key transcription factors represents a transformative advancement in regenerative medicine. Unlike early reprogramming methods that relied on integrating viral vectors, which carried risks of genomic disruption and tumorigenicity, mRNA-based reprogramming offers a non-integrating, "footprint-free" approach [26]. This technology enables the generation of patient-specific pluripotent stem cells without permanent genetic alteration, making it particularly suitable for clinical applications [20]. While fibroblasts have been the traditional cell source for reprogramming studies, emerging research demonstrates that mRNA technology can successfully reprogram diverse somatic cell types, each offering unique advantages for disease modeling and therapeutic development.
The fundamental principle behind mRNA reprogramming involves introducing in vitro transcribed mRNA molecules encoding reprogramming factors into somatic cells. These mRNA molecules are translated into proteins that orchestrate the epigenetic and transcriptional remodeling necessary to achieve a pluripotent state [26]. The most common factors used are the Yamanaka factors (OCT4, SOX2, KLF4, and c-MYC), though variations and supplements to this cocktail have been developed to enhance efficiency and safety [11]. Key advantages of mRNA reprogramming include its high efficiency, precise control over factor stoichiometry and timing, elimination of genomic integration risks, and ability to generate clinical-grade iPSCs [26] [27]. As the field progresses, optimizing this technology for diverse cell sources beyond fibroblasts has become crucial for expanding its research and clinical applications.
Core Principles of mRNA Reprogramming Technology
Molecular Mechanisms and Key Advantages
mRNA reprogramming functions by leveraging the cell's native translational machinery to produce proteins that drive the transition from a somatic to pluripotent state. When synthetic mRNA molecules enter the cell cytoplasm, ribosomes translate them into functional transcription factor proteins that translocate to the nucleus and initiate the reprogramming cascade [26]. These factors activate endogenous pluripotency networks while suppressing somatic cell programs through epigenetic modifications, including changes in DNA methylation and histone acetylation [11]. A critical technical challenge in early mRNA reprogramming was the activation of innate antiviral responses, as cells recognize exogenous RNA through pattern recognition receptors. This obstacle has been overcome through nucleoside modifications (such as pseudouridine substitution) and optimized mRNA cap structures (e.g., using cap1 structures found in higher eukaryotes), which significantly reduce immune activation while enhancing translation efficiency and mRNA stability [28].
The translational control of reprogramming represents another crucial regulatory layer. Research has demonstrated that eukaryotic translation initiation factor 4E (eIF4E) binding proteins (4E-BPs), which are translational repressors, exert multifaceted effects on reprogramming efficiency. Modulating this pathway can influence the translation of endogenous mRNAs such as Sox2 and Myc, significantly impacting the reprogramming process [29]. The flexibility of mRNA technology allows researchers to fine-tune the stoichiometry of reprogramming factors by adjusting the relative concentrations of different mRNA species in the transfection cocktail [26]. This precise control enables optimization for different somatic cell types, which may require varying factor ratios for efficient reprogramming. Furthermore, the transient nature of mRNA expression (typically lasting 24-48 hours per transfection) means that prolonged factor expression, which can inhibit subsequent differentiation, is avoided once the endogenous pluripotency network is established [26] [27].
Comparative Analysis of Reprogramming Delivery Systems
Multiple delivery systems exist for introducing reprogramming factors into somatic cells, each with distinct advantages and limitations. The table below provides a comparative analysis of the primary reprogramming methodologies:
Table 1: Comparison of Key Reprogramming Delivery Systems
| Delivery System | Genomic Integration | Reprogramming Efficiency | Safety Profile | Ease of Use | Best Applications |
|---|---|---|---|---|---|
| Retrovirus/Lentivirus | Yes | Moderate (≈0.01%) | Low (oncogenic risk) | Moderate | Basic research |
| Sendai Virus | No | High | Moderate (persistent viral RNA) | Complex | Research & preclinical |
| Episomal Plasmids | Low frequency | Low | Moderate | Moderate | Research |
| Protein Transduction | No | Very Low | High | Complex | Research |
| mRNA-Based | No | Very High (up to 90.7%) | High | Complex (requires multiple transfections) | Clinical applications |
As evidenced in the table, mRNA reprogramming offers the optimal combination of high efficiency and safety, making it particularly suitable for clinical translation [26] [27]. Unlike DNA-based methods, mRNA does not require nuclear entry for activity and cannot integrate into the host genome. Compared to protein transduction, mRNA reprogramming achieves substantially higher efficiency because the continuous translation from repeated transfections provides sustained intracellular protein levels necessary for the multi-step reprogramming process [26]. While Sendai virus-based systems also offer high efficiency without integration, they involve persistent viral RNA that can be difficult to clear from cultures, creating challenges for clinical applications [26]. The mRNA approach thus represents the most unambiguously transient reprogramming method available.
Technical Framework for mRNA Reprogramming
Essential Reagents and Materials
Successful mRNA reprogramming requires carefully selected reagents and materials optimized for each step of the process. The following table outlines key components of the reprogramming toolkit:
Table 2: Essential Research Reagent Solutions for mRNA Reprogramming
| Reagent Category | Specific Examples | Function & Importance |
|---|---|---|
| Reprogramming mRNAs | Modified mRNAs encoding OCT4, SOX2, KLF4, c-MYC (OSKM); potentially with OCT4 variant (M3O) | Core factors that induce pluripotency; modified nucleosides prevent immune activation |
| Supplemental RNAs | miRNA-367/302s family mimics | Enhance reprogramming efficiency; help overcome epigenetic barriers |
| Transfection Reagent | Lipofectamine RNAiMAX | Efficiently delivers RNA into cells with minimal toxicity |
| Transfection Buffer | Opti-MEM (pH adjusted to 8.2) or PBS | Optimizes transfection efficiency; critical for primary cell reprogramming |
| Culture Medium | Knock-out Serum Replacement (KOSR) medium | Supports cell survival and reprogramming under feeder-free conditions |
| Cell Culture Substrate | Matrigel or recombinant laminin-521 | Provides proper extracellular matrix support for iPSC colony formation and growth |
The synergy between modified mRNAs and supplemental microRNAs significantly enhances reprogramming outcomes. Research demonstrates that combining a 6-factor modified mRNA cocktail (5fM3O) with miRNA-367/302s mimics can increase reprogramming efficiency to unprecedented levels—up to 90.7% of individually plated primary fibroblasts [27]. The optimization of transfection conditions, including buffer pH and composition, proves critical for achieving high transfection efficiency in sensitive primary cell types. Using Opti-MEM adjusted to pH 8.2 as the transfection buffer, for instance, dramatically improves transfection efficiency compared to standard pH buffers [27]. Additionally, low-oxygen culture conditions (approximately 5% O₂) have been shown to facilitate colony formation and improve reprogramming outcomes [28].
Optimized Workflow and Protocol
A highly efficient mRNA reprogramming protocol for human primary cells involves the following key steps:
Cell Preparation and Seeding: Plate 500-1000 human primary somatic cells per well of a 6-well plate in optimized growth medium. Low seeding density allows cells to undergo more cycles of replication, which promotes reprogramming.
Transfection Regimen: Perform transfections every 48 hours using a combination of 600 ng of 5fM3O modified mRNA cocktail and 20 pmol of miRNA-367/302s mimics per well of a 6-well plate. Use Lipofectamine RNAiMAX with Opti-MEM at pH 8.2 as the transfection buffer.
Medium Exchange: Change to fresh reprogramming medium (KOSR-based) 4-6 hours after each transfection to minimize cytotoxicity and maintain cell health.
Colony Monitoring: Observe emerging iPSC colonies approximately 18-25 days after initiating transfections. Colonies typically exhibit sharp borders and high nucleus-to-cytoplasm ratios characteristic of pluripotent stem cells.
Colony Picking and Expansion: Mechanically pick or dissociate distinct colonies and transfer them to fresh culture plates pre-coated with Matrigel or recombinant laminin-521 for expansion under standard human pluripotent stem cell culture conditions.
This protocol requires a minimum of three transfection cycles performed every 48 hours to successfully generate iPSCs, with seven transfections proving optimal for achieving maximum efficiency [27]. Transfections at 72-hour intervals show reduced reprogramming capacity, while intervals shorter than 48 hours increase cytotoxicity without improving efficiency. The entire process from somatic cell to established iPSC line typically takes 4-6 weeks, depending on the source cell type and specific protocol modifications.
Figure 1: The optimized workflow for mRNA reprogramming of primary somatic cells, highlighting critical parameters for success.
Comparative Efficiency Across Cell Types
While fibroblasts remain the most extensively characterized cell source for reprogramming, various somatic cell types present unique advantages for specific applications. The table below summarizes reprogramming outcomes across different cell sources using mRNA technology:
Table 3: mRNA Reprogramming Efficiency Across Diverse Somatic Cell Types
| Cell Source | Reprogramming Efficiency | Time to Colony Emergence | Key Advantages | Notable Challenges |
|---|---|---|---|---|
| Dermal Fibroblasts | Up to 90.7% [27] | 18-25 days | Well-established protocol, accessible tissue source | Invasive biopsy required |
| Peripheral Blood Cells | Moderate to High (data limited) | 21-28 days | Minimal invasion, abundant donor material | Requires specialized culture conditions |
| Adipose-derived Cells | Moderate (data limited) | 20-26 days | Accessible source, high cell yield | Presence of lipid droplets |
| Neural Stem Cells | High (theoretically) | Potentially shorter | Endogenous expression of SOX2 | Limited availability, specialized isolation |
| Urinary Epithelial Cells | Moderate (data limited) | 22-28 days | Non-invasive collection | Potential contamination issues |
The exceptionally high reprogramming efficiency of over 90% was demonstrated specifically with primary neonatal fibroblasts using an optimized mRNA protocol combining modified mRNAs with miRNA-367/302s mimics [27]. This efficiency far surpasses the 0.01% typically achieved with original retroviral methods and represents a significant advancement for the field [26]. Evidence suggests that neural stem cells may demonstrate enhanced reprogramming efficiency due to their endogenous expression of key pluripotency factors like SOX2, potentially reducing the number of exogenous factors required [11]. One study even demonstrated that OCT4 expression alone could successfully generate iPSCs from human neural stem cells, highlighting how the starting cell type influences factor requirements [11].
Cell Type-Specific Methodological Considerations
Different somatic cell sources require tailored approaches for successful mRNA reprogramming:
Blood Cells: Peripheral blood mononuclear cells (PBMCs) represent an attractive cell source due to minimally invasive collection. Successful reprogramming requires pre-activation with cytokines such as IL-2, IL-3, and SCF to promote survival and proliferation before initiating mRNA transfections [20]. T-cells require particular attention to reprogramming factor stoichiometry, as high c-MYC expression can induce apoptosis in these cells.
Adipose-derived Stem Cells (ASCs): These cells naturally express higher levels of mesenchymal stem cell markers and may require adjustments to the standard reprogramming cocktail. The presence of lipid droplets in adipocytes can be mitigated through pre-culture with adipocyte differentiation inhibitors before reprogramming attempts.
Epithelial Cells: Buccal mucosa and urinary epithelial cells can be collected non-invasively but often require careful handling to prevent microbial contamination. These cells typically have faster doubling times than fibroblasts, potentially shortening the reprogramming timeline. Keratinocytes from plucked hair follicles represent another accessible epithelial source that has been successfully reprogrammed using non-mRNA methods, suggesting compatibility with mRNA approaches.
Neural Stem Cells (NSCs): The endogenous expression of SOX2 and other neural progenitor factors in NSCs may allow for reduced factor cocktails. However, obtaining pure NSC populations requires specialized isolation techniques or differentiation from other cell sources, potentially limiting their practical utility for routine reprogramming.
For all cell types, optimizing seeding density represents a critical parameter. The ultra-high efficiency protocol for fibroblasts uses remarkably low seeding densities (500 cells per well of a 6-well plate), which allows individual cells to undergo more divisions—a key facilitator of reprogramming [27]. Other cell types may require density optimization to balance cell-cell contact signaling with proliferation capacity.
Figure 2: Decision framework for selecting appropriate somatic cell sources based on research requirements and practical constraints.
Quality Control and Characterization
Rigorous quality control is essential to ensure the pluripotency, genetic integrity, and differentiation potential of iPSCs generated via mRNA reprogramming. Standard characterization should include:
Pluripotency Marker Expression: Immunofluorescence staining and flow cytometry analysis for surface markers (TRA-1-60, TRA-1-81, SSEA-4) and intracellular transcription factors (OCT4, NANOG, SOX2). The expression of TRA-1-60 is particularly indicative of fully reprogrammed cells [27].
Gene Expression Analysis: RT-PCR or RNA sequencing to verify activation of endogenous pluripotency genes and silencing of the exogenous mRNA reprogramming factors. Complete silencing of the transfected mRNA confirms the establishment of a self-sustaining pluripotency network.
Trilineage Differentiation Capacity: In vitro differentiation through embryonic body formation followed by immunostaining for markers of all three germ layers (ectoderm: βIII-tubulin; mesoderm: α-smooth muscle actin; endoderm: α-fetoprotein). In vivo teratoma formation in immunocompromised mice provides the most stringent test of pluripotency [28].
Karyotype Analysis: G-banding chromosomal analysis to ensure genomic stability after extensive cell divisions during reprogramming and expansion. More comprehensive genomic analysis through whole-genome sequencing can identify copy number variations or point mutations that may have arisen during the process.
Clearance Verification: For clinical applications, verification that no residual reprogramming mRNAs or proteins persist in the established iPSCs. RNA sequencing can confirm the absence of transgenic reprogramming factor expression.
The transient nature of mRNA reprogramming eliminates the need for checking genomic integration of reprogramming factors, simplifying the safety assessment compared to viral methods. However, comprehensive screening should still be performed to rule out potential off-target effects or epigenetic abnormalities that might impact the functionality of differentiated cells derived from the iPSCs.
mRNA reprogramming technology has revolutionized our ability to generate human iPSCs from diverse somatic cell sources with unprecedented efficiency and safety profiles. The optimization of mRNA chemistry, transfection methods, and culture conditions has enabled researchers to overcome previous limitations associated with viral vectors and other non-integrating approaches. While fibroblasts have served as the prototypical cell source, expanding this technology to encompass blood cells, epithelial cells, neural stem cells, and other somatic cell types opens new possibilities for disease modeling, drug screening, and regenerative medicine applications.
Future advancements in mRNA reprogramming will likely focus on further enhancing efficiency across diverse cell types, developing even more refined control over reprogramming factor stoichiometry and timing, and creating closed-system automated platforms for clinical-grade iPSC production. The integration of CRISPR-Cas9 genome editing with mRNA reprogramming presents exciting opportunities for generating genetically corrected patient-specific iPSCs without DNA integration [20]. Additionally, continued optimization of synthetic mRNA formulations and delivery systems may eventually enable in vivo reprogramming approaches, potentially bypassing the need for ex vivo cell culture altogether. As these technologies mature, mRNA-based reprogramming of diverse somatic cell sources will play an increasingly central role in both basic research and clinical applications, bringing us closer to the promise of personalized regenerative medicine.
The convergence of messenger RNA (mRNA) technology with induced pluripotent stem cell (iPSC) research is revolutionizing biomedical science. This whitepaper details how mRNA-driven iPSC generation and differentiation create sophisticated, patient-specific models for studying neurological disorders and cancer. We provide a comprehensive technical guide covering core reprogramming mechanisms, differentiation protocols into neural and immune cell lineages, key experimental workflows, and a reagent toolkit for researchers. The integration of these technologies enables unprecedented precision in disease modeling, drug screening, and the development of next-generation cell therapies.
The discovery that somatic cells could be reprogrammed into induced pluripotent stem cells (iPSCs) represented a paradigm shift in regenerative medicine and disease modeling. Conventional reprogramming methods often relied on viral vectors, raising concerns about insertional mutagenesis and genomic instability [8]. The advent of mRNA-based reprogramming has transformed this landscape by offering a non-integrating, highly efficient alternative for generating clinical-grade iPSCs [8] [11].
This approach involves delivering synthetic mRNA molecules encoding key reprogramming factors, which are translated into proteins within the target cells without ever entering the nucleus or integrating into the host genome [8]. The resulting iPSCs maintain a patient-specific genetic background while achieving a pluripotent state, making them ideal for creating personalized disease models [3]. This technical advance is particularly valuable for studying complex neurological disorders and cancers, where genetic heterogeneity and environmental factors contribute significantly to disease pathogenesis [30].
The versatility of mRNA technology extends beyond initial reprogramming to include directed differentiation into various cell types and genetic engineering of iPSCs using CRISPR-Cas systems [8] [25]. When combined with increasingly complex 3D culture systems, these approaches enable researchers to build sophisticated models that recapitulate key aspects of human tissue architecture and function, providing powerful platforms for investigating disease mechanisms and therapeutic interventions [31] [8].
mRNA Reprogramming Mechanisms and Workflows
Molecular Mechanisms of mRNA-Mediated Reprogramming
The process of reprogramming somatic cells to pluripotency using mRNA involves sophisticated molecular mechanisms that reverse the epigenetic landscape of differentiated cells:
Stochastic Early Phase: The initial delivery of mRNA-encoded reprogramming factors (typically OSKM: OCT4, SOX2, KLF4, c-MYC) triggers a stochastic silencing of somatic genes while partially activating early pluripotency networks. During this phase, the inefficient access of transcription factors to closed chromatin regions creates variability, with only a small fraction of cells progressing to full reprogramming [3].
Deterministic Late Phase: Successful cells enter a more deterministic phase characterized by activation of late pluripotency genes, including NANOG. This transition involves profound epigenomic remodeling, including global DNA demethylation and establishment of pluripotency-specific histone modification patterns [3].
Metabolic Reprogramming: Cells undergoing reprogramming shift from oxidative phosphorylation to glycolysis, mimicking the metabolic state of embryonic stem cells. This metabolic transition supports the biosynthetic demands of rapidly dividing pluripotent cells [3].
Mesenchymal-to-Epithelial Transition (MET): A critical event in reprogramming fibroblasts, MET involves downregulation of mesenchymal markers (SNAI1, SNAI2) and upregulation of epithelial markers (E-cadherin), facilitating the acquisition of pluripotent characteristics [3].
Optimized mRNA Reprogramming Workflow
The following diagram illustrates the standardized workflow for efficient mRNA-mediated somatic cell reprogramming:
Figure 1: mRNA-mediated reprogramming workflow for generating clinical-grade iPSCs from somatic cells.
Key Technical Considerations:
- mRNA Modifications: Incorporation of pseudouridine (ψ) and 5-methylcytidine reduces innate immune recognition and enhances translation efficiency [8].
- Transfection Schedule: Daily transfections for 14-21 days maintain consistent reprogramming factor expression until endogenous pluripotency networks are established.
- Culture Conditions: Defined, xeno-free media systems (e.g., Essential 8 or mTeSR) with vitronectin coating support robust iPSC growth while maintaining compliance with good manufacturing practice (GMP) standards [32].
- Quality Control: Comprehensive characterization includes pluripotency marker assessment (OCT4, SOX2, NANOG, TRA-1-60, SSEA4), karyotyping, and trilineage differentiation potential verification [32].
Neurological Disease Modeling Applications
Differentiation to Neural Lineages
iPSCs generated via mRNA reprogramming can be systematically differentiated into various neural cell types relevant to disease modeling:
Figure 2: Neural differentiation pathways from mRNA-derived iPSCs for neurological disease modeling.
Key Differentiation Protocols:
Motor Neuron Differentiation: Neural progenitor cells are treated with retinoic acid (RA) and sonic hedgehog pathway agonists (SAG) to induce spinal cord identity, followed by maturation with brain-derived neurotrophic factor (BDNF) and glial cell line-derived neurotrophic factor (GDNF) [11]. These iPSC-derived motor neurons (iPSC-MNs) enable modeling of amyotrophic lateral sclerosis (ALS) and related disorders.
Forebrain Neuron Differentiation: Inhibition of WNT and TGF-β signaling pathways directs neural progenitors toward cortical fates, generating glutamatergic neurons for modeling Alzheimer's disease and autism spectrum disorders [30].
Astrocyte Differentiation: Treatment of neural progenitors with ciliary neurotrophic factor (CNTF) and bone morphogenetic protein 4 (BMP4) generates astrocytes for studying neuroinflammatory components of neurological diseases [30].
Disease Modeling and Phenotypic Screening
Patient-derived mRNA-iPSC models recapitulate key pathological features of neurological disorders:
Table 1: Neurological Disease Modeling Using mRNA-iPSC-Derived Cells
| Disease Model | Cell Type Generated | Key Disease Phenotypes | Applications |
|---|---|---|---|
| ALS | Motor neurons | TDP-43 proteinopathy, neurite retraction, reduced neuronal activity, glutamate excitotoxicity [11] | Drug screening, pathway analysis, biomarker identification |
| Parkinson's Disease | Dopaminergic neurons | α-synuclein accumulation, mitochondrial dysfunction, oxidative stress [8] [30] | Neuroprotective compound testing, electrophysiological studies |
| Alzheimer's Disease | Cortical neurons | Aβ42 accumulation, tau hyperphosphorylation, synaptic dysfunction [30] | High-content screening, mechanism studies |
| Lesch-Nyhan Disease | Neural stem cells | Impaired purine recycling, aberrant dopamine signaling [33] | Metabolic pathway analysis, drug toxicity testing |
Advanced Model Systems:
- 3D Organoid Cultures: Self-organizing cerebral organoids containing multiple neuronal subtypes and glial cells model complex cell-cell interactions and tissue-level organization [8].
- Blood-Brain Barrier Models: Co-cultures of iPSC-derived brain microvascular endothelial cells with astrocytes and pericytes enable drug permeability studies [8].
- Microfluidic Platforms: Compartmentalized chambers connected by microgrooves facilitate axon-only treatments and localized injury models [30].
Cancer Research and Immunotherapy Applications
iPSC-Derived Immune Cells for Cancer Immunotherapy
The generation of immune effector cells from mRNA-iPSCs provides a renewable, genetically uniform source for cancer immunotherapy:
Figure 3: Production of engineered immune cells from mRNA-iPSCs for cancer immunotherapy applications.
Key Differentiation Protocols:
Natural Killer (NK) Cell Differentiation: A two-step process involving first the differentiation of iPSCs to CD34+ hematopoietic stem cell-like cells using cytokines and small molecules, followed by NK cell maturation with IL-3, IL-7, IL-15, SCF, and Flt3-L [32]. 3D culture systems significantly improve the yield and functionality of resulting NK cells.
CAR-NK Cell Engineering: CRISPR-Cas9-mediated insertion of chimeric antigen receptors (CARs) targeting tumor-associated antigens (e.g., CD19, BCMA) enhances tumor-specific cytotoxicity [34] [32].
Gamma Delta (γδ) T Cell Generation: Co-culture with Notch ligand-expressing stromal cells or specific cytokine combinations (IL-7, SCF, Flt3-L) drives differentiation toward γδ T cell lineage, creating cells with hybrid NK and T cell recognition capabilities [34].
Enhanced Immunotherapy Through mRNA Vaccine Priming
Recent clinical evidence demonstrates that mRNA-based COVID vaccines generate improved responses to immunotherapy in cancer patients:
Table 2: mRNA Vaccine Enhancement of Cancer Immunotherapy Outcomes
| Cancer Type | Patient Cohort | Intervention | Key Findings | Proposed Mechanism |
|---|---|---|---|---|
| Non-small cell lung cancer | 180 vaccinated vs. 704 unvaccinated patients | mRNA vaccine within 100 days of immune checkpoint inhibition | Median survival: 37.33 months (vaccinated) vs. 20.6 months (unvaccinated) [35] | Vaccine acts as immune alarm, increasing PD-L1 expression on tumors, creating favorable environment for anti-PD-L1 therapy |
| Metastatic melanoma | 43 vaccinated vs. 167 unvaccinated patients | mRNA vaccine with immune checkpoint inhibitors | Significant improvement in median survival (26.67 months unvaccinated, not reached in vaccinated group) [35] | Training of immune system to eliminate cancer cells regardless of vaccine antigen specificity |
| Immunologically "cold" tumors | Patients with low PD-L1 expression | mRNA vaccine with immunotherapy | Nearly five-fold improvement in three-year overall survival [35] | Conversion of immunologically inactive tumors to responsive state |
Mechanistic Insights:
The observed synergy between mRNA vaccines and cancer immunotherapy appears to involve:
- Innate Immune Activation: mRNA vaccines stimulate pattern recognition receptors (TLRs, RIG-I), producing a generalized state of immune alertness that enhances responses against tumor antigens [35].
- PD-L1 Upregulation: Vaccine-induced immune activation triggers interferon signaling, leading to increased PD-L1 expression on tumor cells, making them more susceptible to PD-1/PD-L1 checkpoint blockade [35].
- Epitope Spreading: The broad immune activation enables recognition of tumor neoantigens beyond the original vaccine target, overcoming tumor immune evasion mechanisms [35].
Advanced Gene Editing and GMP Compliance
CRISPR-Cas Gene Editing in mRNA-iPSCs
Precise genetic manipulation of mRNA-derived iPSCs enables creation of sophisticated disease models and enhancement of therapeutic cells:
Sequential Factor Delivery Protocol for Efficient Knock-in:
- Day 0 - Donor Template Delivery: Nucleofect 3×10^6 iPSCs with donor plasmid DNA using P4 Nucleofection Buffer and program CA-167 [25].
- Recovery: Incubate cells in RPMI medium for 10 minutes post-nucleofection to significantly improve cell survival [25].
- Day 1 - RNP Complex Delivery: Nucleofect cells with pre-complexed Cas9/Cas12a ribonucleoproteins (RNPs) targeting desired genomic locus [25].
- Cold Shock Incubation: Maintain cells at 32°C for 24-48 hours to enhance homology-directed repair (HDR) efficiency [25].
- Clone Isolation: Seed cells by limiting dilution into 96-well plates 5-7 days post-editing [25].
- Screening: Identify biallelically edited clones via flow cytometry or PCR-based genotyping [25].
This optimized approach achieves knock-in efficiencies of up to 40% without requiring antibiotic selection or complex instrumentation, maintaining compatibility with GMP standards [25].
Safety Engineering for Therapeutic Applications
Critical genetic modifications to enhance safety and efficacy of iPSC-derived therapies:
- Inducible Safety Switches: Integration of inducible caspase-9 (iCaspase9) genes enables elimination of engineered cells in case of adverse events through administration of a small molecule dimerizer [25].
- HLA Engineering: Knock-out of beta-2-microglobulin (B2M) ablates surface expression of HLA class I molecules, creating universally compatible "off-the-shelf" cell products that evade host T-cell recognition [34] [25].
- Suicide Gene Systems: Incorporation of herpes simplex virus thymidine kinase (HSV-TK) allows ganciclovir-mediated ablation of proliferating cells, providing a safeguard against potential tumor formation [25].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for mRNA-iPSC Workflows
| Reagent Category | Specific Products | Application | Technical Notes |
|---|---|---|---|
| Reprogramming mRNA | Stemgent mRNA Reprogramming Cocktail | Footprint-free iPSC generation | Includes modified OSKM mRNAs with pseudouridine for reduced immunogenicity [8] |
| Cell Culture Media | Essential 8 Medium, mTeSR Plus | iPSC maintenance and expansion | Defined, xeno-free formulations supporting pluripotency [32] |
| Extracellular Matrices | Vitronectin, Recombinant Laminin-521 | iPSC attachment and growth | Chemically defined alternatives to Matrigel for GMP compliance [32] |
| Neural Differentiation | STEMdiff Neural System | Neural induction and differentiation | Dual SMAD inhibition protocol for efficient neural conversion [11] |
| Hematopoietic Cytokines | IL-3, IL-7, IL-15, SCF, Flt3-L | Immune cell differentiation | Critical for NK cell and T cell development from iPSCs [32] |
| Gene Editing Tools | Alt-R S.p. HiFi Cas9, Alt-R A.s. Cas12a | CRISPR genome editing | High-fidelity nucleases with reduced off-target effects [25] |
| Transfection Reagents | Lipofectamine MessengerMAX, Ribojuice | mRNA delivery | Optimized for high-efficiency mRNA transfection with low cytotoxicity [31] [8] |
| 3D Culture Systems | Corning Matrigel, Synthemax | Spheroid and organoid culture | Supports complex 3D structures for advanced disease modeling [31] |
The integration of mRNA technology with iPSC-based disease modeling represents a transformative approach for studying neurological disorders and cancer. The non-integrating nature of mRNA reprogramming combined with precise CRISPR genome editing enables generation of genetically defined, patient-specific models that recapitulate disease pathophysiology with unprecedented fidelity. Standardized differentiation protocols yield functional neural and immune cells for mechanistic studies and therapeutic development, while advanced 3D culture systems capture the complexity of human tissues. As these technologies continue to mature, they promise to accelerate drug discovery and enable development of safer, more effective cell therapies for conditions that have traditionally proven difficult to model and treat.
The convergence of induced pluripotent stem cell (iPSC) and messenger RNA (mRNA) technologies represents a paradigm shift in biomedical research and therapeutic development. mRNA-based reprogramming offers a non-integrating, highly controllable method for generating patient-specific iPSCs, effectively addressing critical safety concerns associated with viral vectors. This whitepaper examines the integral role of mRNA technology in advancing iPSC applications from foundational research to clinical implementation. We provide a technical analysis of how mRNA-engineered iPSCs are revolutionizing drug discovery through physiologically relevant disease models and high-throughput screening platforms, while simultaneously paving the way for personalized regenerative therapies. The integration of these platforms enables unprecedented precision in modeling human diseases, screening therapeutic compounds, and developing patient-specific treatments, fundamentally transforming the pipeline from basic research to clinical application.
The discovery that somatic cells could be reprogrammed into induced pluripotent stem cells (iPSCs) using defined transcription factors marked a transformative milestone in regenerative medicine [20] [36]. Shinya Yamanaka's pioneering work identified OCT4, SOX2, KLF4, and c-MYC (OSKM) as key factors capable of resetting cellular epigenetics to a pluripotent state [11] [19]. While this breakthrough opened unprecedented opportunities for disease modeling and cell-based therapies, early reprogramming methodologies relying on viral vector integration raised significant safety concerns regarding insertional mutagenesis and tumorigenic potential [20] [37].
The emergence of mRNA-based reprogramming has provided a revolutionary solution to these challenges. Unlike traditional viral methods, mRNA technology enables transient expression of reprogramming factors without genomic integration, significantly enhancing the safety profile of derived iPSCs [9] [36]. This non-integrating approach maintains high reprogramming efficiency while eliminating the risk of permanent genetic alterations, making it particularly suitable for clinical applications [37]. Furthermore, mRNA platforms offer precise control over factor expression timing and dosage, allowing optimized reprogramming kinetics and enhanced reproducibility [9].
The versatility of mRNA technology extends beyond initial reprogramming to include directed differentiation and functional maturation of iPSC-derived lineages. Advances in nucleotide chemistry and delivery systems have substantially improved mRNA stability and translation efficiency while reducing immunogenicity, establishing mRNA as a powerful tool for controlling cell fate and function [9]. These developments have positioned mRNA-engineered iPSCs as a cornerstone technology for both drug screening platforms and personalized regenerative medicine applications.
Technical Foundations: mRNA-Based iPSC Generation and Characterization
mRNA Reprogramming Methodology
The generation of iPSCs using mRNA technology requires meticulous optimization of multiple parameters to achieve efficient reprogramming while maintaining cell viability. The following protocol outlines a standardized approach for mRNA-based reprogramming of human somatic cells:
Cell Preparation: Plate human dermal fibroblasts or peripheral blood mononuclear cells (PBMCs) in appropriate culture vessels at a density of 10,000-50,000 cells/cm². Maintain cells in complete medium for 24-48 hours until they reach 70-80% confluence [19].
mRNA Formulation: Prepare a cocktail of synthetic mRNA molecules encoding OCT4, SOX2, KLF4, c-MYC, and LIN28. Incorporate modified nucleotides (e.g., pseudouridine) to reduce innate immune recognition and enhance translational efficiency [9]. Include a traceable fluorescent tag for transfection efficiency monitoring.
Transfection Protocol: Complex mRNA with a transfection reagent (e.g., lipid nanoparticles or polymer-based carriers). For each well of a 6-well plate, combine 1-2 µg of total mRNA with appropriate transfection reagent in serum-free medium. Incubate for 15-20 minutes at room temperature to form complexes [9] [19].
Reprogramming Cycle: Aspire culture medium from prepared cells and add mRNA-transfection complexes. Incubate cells for 4-6 hours at 37°C, 5% CO₂, then replace with fresh complete medium. Repeat transfections daily for 16-21 days with medium changes between transfections [19].
Colony Selection and Expansion: Monitor cultures for emergence of compact, ESC-like colonies with defined borders between days 18-25. Mechanically pick individual colonies and transfer to feeder-free culture conditions using defined essential 8 medium or mTeSR. Expand and characterize clones for pluripotency marker expression and differentiation potential [19].
Table 1: Key Research Reagents for mRNA-Based iPSC Generation
| Reagent Category | Specific Product/Component | Function in Reprogramming |
|---|---|---|
| Reprogramming Factors | OCT4, SOX2, KLF4, c-MYC mRNA | Core transcription factors for epigenetic reprogramming [11] |
| Supplemental Factors | LIN28, NANOG mRNA | Enhances reprogramming efficiency and kinetics [19] |
| Nucleotide Modifications | Pseudouridine, 5-methylcytidine | Reduces immunogenicity, increases mRNA stability and translation [9] |
| Delivery System | Lipid nanoparticles (LNPs) | Enables efficient cellular mRNA uptake and endosomal escape [9] |
| Culture Medium | Essential 8, mTeSR | Defined, xeno-free medium supporting pluripotency maintenance [19] |
| Small Molecules | Valproic acid, CHIR99021 | Epigenetic modulators that enhance reprogramming efficiency [36] |
Characterization of mRNA-Derived iPSCs
Rigorous quality control is essential to confirm successful reprogramming and pluripotency of mRNA-derived iPSCs. The following characterization pipeline ensures the generation of high-quality iPSC lines:
Morphological Assessment: Daily observation for emergence of embryonic stem cell-like colonies with high nucleus-to-cytoplasm ratio, prominent nucleoli, and compact colony formation with defined borders [19].
Pluripotency Marker Verification: Immunofluorescence staining and flow cytometry analysis for key pluripotency markers including OCT4, SOX2, NANOG, SSEA-4, TRA-1-60, and TRA-1-81 [38] [19]. Quantitative PCR for endogenous expression of pluripotency genes.
Trilineage Differentiation Potential: In vitro differentiation through embryonic body formation followed by immunostaining for ectodermal (β-III-tubulin, PAX6), mesodermal (α-smooth muscle actin, brachyury), and endodermal (α-fetoprotein, SOX17) markers [38] [19].
Karyotype Analysis: G-banding chromosomal analysis at passage 5-10 to confirm genomic integrity and absence of major chromosomal abnormalities [38].
Identity Confirmation: Short tandem repeat (STR) profiling to verify donor cell line matching and exclude cross-contamination [39].
The following diagram illustrates the complete workflow for mRNA-based iPSC generation and characterization:
Diagram 1: mRNA-based iPSC generation and characterization workflow
iPSC Platforms in Drug Discovery and Development
Disease Modeling and High-Throughput Screening
iPSC technology has revolutionized pharmaceutical research by enabling the generation of human disease models with unprecedented physiological relevance. Patient-derived iPSCs retain the complete genetic background of the donor, including disease-associated mutations and polymorphisms, allowing for the recapitulation of disease phenotypes in vitro [37] [39]. This approach is particularly valuable for neurological disorders, cardiac conditions, and rare genetic diseases where animal models often fail to predict human therapeutic responses [39].
A landmark study demonstrating the power of iPSC-based disease modeling involved the creation of an iPSC library from 100 sporadic amyotrophic lateral sclerosis (SALS) patients [39]. This library enabled large-scale phenotypic screening and identification of therapeutic candidates effective across a genetically heterogeneous patient population. The motor neurons derived from these SALS-iPSCs recapitulated key disease pathologies including reduced survival, accelerated neurite degeneration, and transcriptional dysregulation [39]. Importantly, this model demonstrated pharmacological rescue by riluzole, validating its clinical relevance [39].
The integration of mRNA technology further enhances these disease models by facilitating the efficient differentiation of iPSCs into relevant cell types. mRNA-based expression of transcription factors and morphogens enables precise control over differentiation pathways without genomic integration, producing more physiologically authentic models for compound screening [9].
Table 2: Applications of iPSC Technology in Drug Discovery
| Application Area | Key Advantages | Representative Example |
|---|---|---|
| Disease Modeling | Patient-specific genetic background; Recapitulation of human pathophysiology | ALS motor neurons showing disease-specific degeneration [39] |
| High-Throughput Screening | Human genetic context; Cell-type specific responses | Screening of 100+ ALS trial drugs identified 3 effective compounds [39] |
| Toxicity Assessment | Human-relevant toxicity profiles; Prediction of clinical cardiotoxicity | iPSC-derived cardiomyocytes for arrhythmia risk assessment [20] |
| Personalized Medicine | Individual drug response variability; Patient stratification | iPSC-derived neurons predicting individual responses to therapies [37] |
| Drug Repurposing | Identification of novel indications for existing drugs | Statins identified as potential therapy for achondroplasia [37] |
Experimental Protocol: Large-Scale Drug Screening in iPSC-Derived Motor Neurons
The following protocol outlines the methodology for large-scale drug screening using iPSC-derived motor neurons, as demonstrated in sporadic ALS research [39]:
iPSC Library Generation:
- Generate iPSCs from 100 SALS patients and 25 healthy controls using non-integrating episomal vectors.
- Subject all lines to rigorous quality control including genomic integrity assessment, pluripotency verification, and trilineage differentiation potential.
- Perform whole-genome sequencing to identify pathogenic variants and confirm sporadic disease status.
Motor Neuron Differentiation:
- Adapt a five-stage spinal motor neuron differentiation protocol with extensively optimized maturation conditions.
- Culture cells for 50-60 days to achieve mature motor neuron phenotypes.
- Verify motor neuron purity (≥92%) via immunostaining for ChAT, MNX1/HB9, and Tuj1.
- Confirm minimal contamination from astrocytes (<0.12%) and microglia (<0.04%).
Phenotypic Screening Platform:
- Implement longitudinal live-cell imaging with motor neuron-specific HB9-turboGFP reporter.
- Monitor cultures daily for survival and neurite integrity.
- Establish quantitative metrics for motor neuron health and degeneration kinetics.
Compound Library Screening:
- Test drugs previously evaluated in ALS clinical trials (n>100) across the SALS motor neuron library.
- Include riluzole as positive control for pharmacological rescue.
- Use concentration-response curves for efficacy assessment.
Combinatorial Therapy Assessment:
- Test effective single agents in combination to identify synergistic effects.
- Validate promising combinations across the entire SALS donor spectrum.
- Assess transcriptomic and electrophysiological rescue in addition to survival benefits.
This approach identified that less than 5% of drugs tested in ALS clinical trials demonstrated efficacy in the human iPSC-derived model, accurately reflecting clinical trial outcomes [39]. Furthermore, combinatorial testing revealed baricitinib, memantine, and riluzole as a promising therapeutic combination for SALS, demonstrating the power of iPSC-based platforms for identifying effective treatment strategies [39].
The screening workflow is visualized in the following diagram:
Diagram 2: Large-scale drug screening workflow using iPSC-derived motor neurons
iPSC-Based Regenerative Medicine and Clinical Translation
Clinical Applications and Trial Outcomes
The therapeutic potential of iPSC-derived cells is being actively investigated across multiple clinical domains, with notable advancements in neurodegenerative diseases, retinal disorders, and cardiac conditions. The non-integrating nature of mRNA reprogramming makes it particularly suitable for generating clinical-grade iPSCs for these applications [9] [36].
Recent clinical trials demonstrate steady progress in iPSC-based therapies:
Parkinson's Disease: A Phase I/II trial (jRCT2090220384) reported in 2025 that allogeneic iPSC-derived dopaminergic progenitors survived transplantation, produced dopamine, and did not form tumors in Parkinson's patients [20] [36]. Concurrently, an ongoing autologous iPSC-derived dopamine neuron trial at Mass General Brigham is pioneering the use of a patient's own blood-derived iPSCs, eliminating the need for immune suppression [20] [36].
Retinal Disorders: Eyecyte-RPE, an iPSC-derived retinal pigment epithelium product, received IND approval in India in 2024 for geographic atrophy associated with age-related macular degeneration [20] [36]. This represents a significant step toward scalable and cost-effective cell therapy approaches.
Cardiac Repair: Preclinical studies in non-human primates demonstrate that iPSC-derived cardiomyocyte patches improve cardiac performance, though transient arrhythmias indicate ongoing safety challenges that require further optimization [20] [36].
Manufacturing and Safety Considerations
The clinical translation of iPSC technologies requires stringent manufacturing protocols and comprehensive safety assessments:
GMP-Compliant Production: Implementation of automated robotics platforms for reprogramming and differentiation to maximize output and uniformity [39]. Use of xenogeneic-free culture systems and closed-system bioreactors for large-scale expansion.
Genomic Stability Monitoring: Regular karyotyping and whole-genome sequencing to detect potential mutations acquired during reprogramming or culture expansion [20] [19].
Tumorigenicity Risk Mitigation: Rigorous purification of differentiated cell products to eliminate residual undifferentiated iPSCs [20]. Extensive pre-clinical teratoma formation assays in immunocompromised mice.
Immunogenicity Management: Use of autologous iPSCs or HLA-matched allogeneic banks to minimize immune rejection [36]. The Kyoto University iPSC Research and Application Center is developing a bank where 75 lines could cover 80% of the Japanese population through HLA matching [19].
Table 3: Clinical Trials and Applications of iPSC-Based Therapies
| Therapeutic Area | Cell Type | Clinical Stage | Key Findings |
|---|---|---|---|
| Parkinson's Disease | Dopaminergic progenitors | Phase I/II Trial | Cell survival, dopamine production, no tumor formation [20] [36] |
| Age-Related Macular Degeneration | Retinal Pigment Epithelium | IND Approval (India) | Step toward scalable therapy; ongoing safety assessment [20] [36] |
| Cardiac Repair | Cardiomyocyte patches | Preclinical (Non-human primates) | Improved cardiac function with transient arrhythmias [20] [36] |
| Osteoarthritis | Mesenchymal stem cells | Preclinical (Arthritic rat model) | Improved cartilage damage, persistence in joint cavity [40] |
| Neurological Disorders | Neural progenitor cells | Disease modeling | Diminished differentiation capacity in DNA repair deficiency [38] |
Emerging Trends and Future Perspectives
The continued evolution of iPSC technology is being shaped by several converging innovations that promise to enhance both research and clinical applications:
Advanced Delivery Platforms: Next-generation mRNA delivery systems utilizing optimized lipid nanoparticles and biomaterial scaffolds are improving the efficiency and specificity of reprogramming and differentiation [9]. These platforms enable spatially and temporally controlled expression of multiple transcription factors, mimicking developmental signaling gradients.
CRISPR/iPSC Integration: The combination of CRISPR-Cas9 genome editing with iPSC technology enables precise genetic correction of disease-associated mutations in patient-specific cells [20] [36]. This approach has been successfully employed to correct mutations in Parkinson's disease patients' iPSCs, resulting in improved mitochondrial function and neuronal integrity [20] [36].
Complex Model Systems: Development of 3D organoid systems and organ-on-chip technologies incorporating multiple cell types derived from iPSCs provides more physiologically relevant models for disease studying and drug screening [37]. These systems better recapitulate tissue-level organization and cell-cell interactions crucial for human pathophysiology.
AI-Guided Differentiation: Machine learning algorithms are being applied to optimize differentiation protocols, predict lineage specification outcomes, and enhance quality control in iPSC manufacturing [20] [36]. These approaches improve reproducibility and standardization of iPSC-derived products.
Extracellular Vesicle Therapeutics: iPSC-derived extracellular vesicles (EVs) are emerging as a cell-free alternative for regenerative applications, offering improved safety profiles while maintaining therapeutic effects [40]. EVs from iPSC-derived mesenchymal stem cells maintain their anti-inflammatory properties longer than those from conventional MSCs [40].
As these technologies mature, they will further accelerate the translation of iPSC-based applications from bench to bedside, ultimately fulfilling the promise of personalized regenerative medicine and transforming the drug development landscape.
The integration of mRNA and iPSC technologies has created a powerful platform that is fundamentally reshaping the landscape of drug discovery and regenerative medicine. mRNA-based reprogramming addresses critical safety concerns associated with earlier methodologies while providing precise control over cell fate specification. The ability to generate patient-specific iPSCs and differentiate them into disease-relevant cell types enables unprecedented opportunities for modeling human disorders, screening therapeutic compounds, and developing personalized cell therapies.
As demonstrated by successful applications in disease modeling, high-throughput drug screening, and early-stage clinical trials, these technologies are bridging the gap between preclinical research and clinical application. The continued refinement of mRNA delivery systems, differentiation protocols, and manufacturing processes will further enhance the safety, efficacy, and scalability of iPSC-based approaches. Through these advancements, the vision of truly personalized medicine—with patient-specific disease models and individually tailored regenerative therapies—is progressively becoming a clinical reality.
Navigating Technical Hurdles: Strategies for Enhancing mRNA Reprogramming Efficiency and Safety
The application of messenger RNA (mRNA) technology in induced pluripotent stem cell (iPSC) research represents a transformative approach for cellular reprogramming and differentiation. However, a significant technical challenge impedes its efficiency: the innate immune system's robust recognition of exogenous mRNA, triggering a potent type I interferon (IFN-I) response. This response, while central to antiviral defense, can be highly detrimental to iPSC generation and function, leading to reduced reprogramming efficiency, impaired cell viability, and disrupted differentiation potential. Recent research has revealed that pluripotency and the canonical IFN-I system are functionally incompatible; inducing antiviral defenses in iPSCs dysregulates pluripotency markers and compromises their ability to form normal ectoderm, endoderm, and mesodermal lineages [41]. This whitepaper provides an in-depth technical analysis of the mechanisms underlying mRNA-sensed immune activation and outlines validated experimental strategies to mitigate this response, thereby enhancing the precision and efficacy of mRNA-based applications in iPSC research.
Molecular Mechanisms of mRNA-Sensed Immune Activation
Understanding the pathways through which exogenous mRNA activates the innate immune system is crucial for developing effective countermeasures. The core issue is that transfected mRNA is recognized by various pattern recognition receptors (PRRs) as a pathogen-associated molecular pattern (PAMP).
Key Sensing Pathways and Downstream Signaling
- Cytosolic Sensors (RIG-I and MDA5): These receptors detect viral RNA in the cytoplasm. RIG-I is particularly adept at recognizing RNA with 5'-triphosphates, while MDA5 senses long double-stranded RNA (dsRNA) structures. Upon activation, they initiate a signaling cascade that culminates in the production of IFN-I [42].
- Endosomal Sensors (TLR7 and TLR8): These Toll-like receptors within endosomal compartments can detect single-stranded RNA (ssRNA) from degraded mRNA cargo, leading to a separate signaling pathway that also induces IFN-I and pro-inflammatory cytokines [42].
- Downstream Inflammatory Response: The signaling cascades from these PRRs converge on the activation of transcription factors such as IRF3 and IRF7, driving the expression of interferon-beta (IFN-β) and other interferon-stimulated genes (ISGs). The secreted IFN-β then binds to the interferon-α/β receptor (IFNAR) on neighboring cells, activating the JAK-STAT pathway and establishing an antiviral state characterized by the widespread expression of hundreds of ISGs [43] [42].
This IFNAR-dependent response is a major barrier to efficient mRNA-based reprogramming. A 2025 study demonstrated that the mRNA component itself—rather than the delivery vehicle—is essential for inducing this potent innate immune activation, which subsequently attenuates the desired adaptive immune response or cellular reprogramming process [43] [42].
Table 1: Key Innate Immune Sensors for Exogenous mRNA
| Sensor | Location | Ligand | Downstream Effect |
|---|---|---|---|
| RIG-I | Cytoplasm | RNA with 5'-triphosphates, short dsRNA | IRF3/7 activation, IFN-β production |
| MDA5 | Cytoplasm | Long dsRNA | IRF3/7 activation, IFN-β production |
| TLR7/TLR8 | Endosome | Single-stranded RNA (ssRNA) | Pro-inflammatory cytokine production |
The Fundamental Incompatibility of Pluripotency and IFN Signaling
A critical consideration for iPSC researchers is the inherent conflict between the IFN-I system and the maintenance of pluripotency. Somatic cells possess a robust IFN-I system, but during reprogramming to pluripotency, this system is largely silenced. Research has shown that iPSCs not only fail to induce IFN-β in response to viral infection or dsRNA but also remain unresponsive to exogenous IFN-β treatment [41].
The molecular basis for this incompatibility lies in the core reprogramming factors. The expression of factors like KLF4 during reprogramming can directly repress the induction of canonical antiviral pathways. Forcing the engagement of antiviral defenses in iPSCs, even transiently, dysregulates genes associated with all three germ layers and compromises subsequent differentiation, particularly affecting ectoderm and endoderm formation [41]. Therefore, uncontrolled IFN activation during the mRNA-based generation of iPSCs can fundamentally undermine their quality and utility.
Experimental Strategies to Mitigate the Interferon Response
Several well-validated methodological approaches can be employed to circumvent or suppress the innate immune response to transfected mRNA, thereby improving outcomes in iPSC research.
mRNA Engineering and Purification
The most proactive strategy involves modifying the mRNA molecule itself to evade detection by PRRs.
- Nucleoside Modification: Replacing uridine with naturally occurring analogs like N1-methylpseudouridine (m1Ψ) is a cornerstone of modern mRNA technology. This modification creates "immuno-silent" mRNA by reducing its affinity for TLR7/8 and other RNA sensors, thereby minimizing IFN induction while simultaneously enhancing translation efficiency and stability [42].
- Removal of dsRNA Impurities: Even in vitro-transcribed mRNA can contain dsRNA contaminants, a potent PAMP. Using cellulose-based purification or HPLC to remove these dsRNA species is a critical step. Studies confirm that this purification dramatically reduces innate immune activation and increases protein yield [42].
- Cap and Tail Engineering: Employing a synthetic Cap 1 structure (e.g., CleanCap) instead of a Cap 0 structure helps evade detection by IFIT sensors. Furthermore, optimizing the 3' poly(A) tail length contributes to mRNA stability and translational efficiency.
Table 2: Summary of Key Mitigation Strategies and Their Mechanisms
| Strategy Category | Specific Method | Mechanism of Action | Key Considerations |
|---|---|---|---|
| mRNA Design | Nucleoside modification (e.g., m1Ψ) | Evades detection by TLR7/8 and other sensors; enhances translation | Foundation of modern mRNA tech |
| Removal of dsRNA impurities | Eliminates potent PAMP recognized by MDA5/RIG-I | Critical purification step post-IVT | |
| Cap 1 structure | Prevents detection by IFIT family proteins | Standard in commercial kits | |
| Pharmacologic Inhibition | Small-molecule JAK inhibitors (e.g., Deucravacitinib) | Blocks IFNAR/JAK-STAT downstream signaling | Transient use is often sufficient |
| Anti-IFNAR antibody | Physically blocks IFN-I binding and signaling | Used in vivo in model systems | |
| Alternative Methods | Sendai virus vector | Non-integrating, cytoplasmic replication | High efficiency, requires clearance |
| mRNA transfection in defined conditions | Optimized timing and cell state for delivery | Reduces peak IFN exposure |
Pharmacological Inhibition of IFN Signaling
When the use of unmodified or minimally modified mRNA is necessary, transient pharmacological inhibition of the IFN signaling pathway can be highly effective.
- Small-Molecule JAK Inhibitors: Compounds like Deucravacitinib inhibit tyrosine kinases (JAKs) downstream of IFNAR, preventing the phosphorylation of STAT proteins and the transcription of ISGs. A typical protocol involves treating cells with a low micromolar concentration (e.g., 1-10 µM) of the inhibitor during and for 24-48 hours after mRNA transfection [42].
- Anti-IFNAR Antibodies: For in vivo applications, a potent strategy is the direct blockade of the IFNAR receptor. In murine studies, intraperitoneal injection of 2.5 mg of an anti-IFNAR monoclonal antibody 24 hours before and 24 hours after immunization with LNP-mRNA successfully abrogated the innate immune response and enhanced subsequent adaptive immunity [43] [42]. This demonstrates that even brief IFNAR blockade can be sufficient to improve outcomes.
Selection of Reprogramming Methodologies
The choice of how to deliver reprogramming factors is paramount. While synthetic mRNA offers speed and efficiency, its immune-activating potential must be managed. Alternatively, non-integrating viral vectors, such as the Sendai virus, provide a highly efficient and widely adopted method for generating clinical-grade iPSCs. The Sendai virus is a cytoplasmic RNA virus that does not integrate into the host genome and is eventually diluted out over cell passages, making it a safe option for clinical applications [8]. This method inherently bypasses some of the toll-like receptor sensing that plagues synthetic mRNA transfection.
Technical Protocols and Workflows
This section outlines detailed experimental protocols for key strategies discussed above, providing a ready-to-use resource for laboratory implementation.
Protocol: Transient IFNAR Blockade for Enhanced mRNA Transfection
This protocol is adapted from studies on LNP-mRNA vaccines and can be modified for iPSC reprogramming or differentiation experiments [43] [42].
Materials:
- Anti-IFNAR monoclonal antibody (e.g., Leinco Technologies, I-401-100)
- mRNA of interest (e.g., modified mRNA encoding reprogramming factors)
- Appropriate transfection reagent (e.g., lipid nanoparticles, polymer-based)
- Cell culture media and supplements
Procedure:
- Pre-treatment: 24 hours prior to mRNA transfection, add the anti-IFNAR antibody to the cell culture medium at a final concentration sufficient to achieve receptor saturation (e.g., 10 µg/mL for in vitro applications).
- Transfection: Perform the mRNA transfection according to established protocols for your cell type.
- Post-treatment: Maintain the antibody in the culture medium for 24 hours post-transfection.
- Assessment: Refresh the medium with standard growth media. Evaluate transfection efficiency, cell viability, and IFN response markers (e.g., MX1 expression by qPCR or phospho-STAT1 by Western blot) 48-72 hours post-transfection.
Protocol: In Vitro Evaluation of IFN Response Using iPSC-Derived Models
iPSC-derived dendritic cells (DCs) provide a robust human cell model for studying IFN pathology and screening inhibitors [44].
Differentiation of Dendritic Cells from iPSCs:
- Hematopoietic Progenitor Cell (HPC) Generation: Detach iPSC colonies and seed them on a layer of irradiated C3H10T1/2 feeder cells in hematopoietic differentiation medium supplemented with VEGF, Flt-3L, SCF, and TPO.
- Harvest HPCs: On day 14, harvest floating hematopoietic progenitor cells from the "sac-like" structures that form.
- DC Differentiation: Re-seed the HPCs onto fresh feeder cells with a cytokine cocktail (Flt-3L, SCF, TPO) to promote myeloid and specifically plasmacytoid DC differentiation, characterized by surface markers CD123 and CD303.
- Maturation and Assay: Around day 21-25, harvest the iPSC-derived DCs and use them for assays. To test IFN inhibitors, add candidate compounds on day 23 and collect supernatants and cells on day 25 for ELISA (IFN-α secretion), qPCR (ISG expression), and functional assays [44].
Diagram 1: Workflow for differentiating iPSCs into DCs for interferon response testing.
Table 3: Key Research Reagent Solutions for Interferon Response Research
| Reagent / Tool | Function/Description | Example Use Case |
|---|---|---|
| N1-methylpseudouridine (m1Ψ) | Modified nucleoside for synthesizing "immuno-silent" mRNA | Evading TLR7/8 sensing in mRNA reprogramming kits [42] |
| Anti-IFNAR antibody | Monoclonal antibody for blocking the type I interferon receptor | Transiently inhibiting IFN signaling in vivo or in complex cell cultures [43] |
| JAK Inhibitor (e.g., Deucravacitinib) | Small molecule inhibitor of JAK/STAT signaling pathway | Suppressing downstream ISG expression post-mRNA delivery [42] |
| Sendai Virus Vectors | Non-integrating, cytoplasmic RNA viral vector for gene delivery | Footprint-free delivery of OSKM factors for iPSC generation [8] |
| CRISPR/Cas9 System | Precision genome editing tool | Generating IFN-pathway reporter lines or knockout models (e.g., IFIH1 R779H) [44] [45] |
| Poly(I:C) | Synthetic analog of dsRNA; potent inducer of IFN-I | Positive control for stimulating the MDA5/RIG-I pathway in assay development [45] |
The mitigation of the innate immune response to mRNA is not merely a technical optimization but a fundamental requirement for unlocking the full potential of mRNA technology in iPSC research and therapy. The strategies outlined here—from biochemical refinement of the mRNA molecule itself to the strategic use of transient immunosuppression—provide a comprehensive toolkit for researchers to enhance reprogramming efficiency, differentiation fidelity, and the overall success of iPSC-based applications. As the field advances, future work will likely focus on even more precise temporal control over immune modulation and the development of novel engineered systems, such as the doxycycline-inducible IFN-β iPSC model [45], which allow for the controlled study of interferon effects. By systematically addressing the challenge of interferon activation, scientists can continue to leverage the remarkable power of mRNA to drive innovations in regenerative medicine, disease modeling, and cell-based therapies.
Optimizing Reprogramming Efficiency through mRNA Modifications and Delivery Systems
The generation of induced pluripotent stem cells (iPSCs) represents a transformative approach in regenerative medicine and disease modeling. However, the clinical translation of iPSC technology remains hampered by the low efficiency of somatic cell reprogramming. This whitepaper examines cutting-edge strategies to enhance reprogramming efficiency through optimized mRNA design and advanced delivery systems. By leveraging chemically modified mRNAs, optimized transfection protocols, and novel non-viral delivery platforms, researchers can achieve unprecedented reprogramming efficiencies exceeding 90% in primary human fibroblasts. These advancements provide a robust, integration-free foundation for producing clinically relevant iPSCs, accelerating their therapeutic application while maintaining compliance with Good Manufacturing Practice (GMP) standards.
Induced pluripotent stem cells (iPSCs) hold tremendous promise for regenerative medicine, disease modeling, and drug discovery due to their capacity for unlimited self-renewal and differentiation into virtually any cell type. Among the various methods for generating iPSCs, mRNA-based reprogramming has emerged as particularly advantageous for clinical applications because it is integration-free, eliminating the risk of insertional mutagenesis associated with viral vectors [8]. This approach involves introducing synthetic modified mRNAs (mod-mRNAs) encoding the essential reprogramming factors (OCT4, SOX2, KLF4, c-MYC, and occasionally LIN28 or NANOG) into somatic cells, transiently expressing these factors to initiate pluripotency induction [27].
Despite its theoretical advantages, mRNA-based reprogramming faces significant challenges including low efficiency, activation of innate immune responses, and difficulties in transfecting certain primary cell types [27]. The transient nature of mRNA expression requires repeated transfections, which can induce cytotoxicity and impede the reprogramming process. Furthermore, different cell types exhibit varying susceptibility to RNA transfection, necessitating optimized delivery strategies tailored to specific starting materials. This technical guide addresses these challenges by presenting the latest advancements in mRNA modifications, delivery systems, and experimental protocols designed to maximize reprogramming efficiency while maintaining cell viability and ensuring the clinical relevance of the resulting iPSCs.
Key mRNA Modification Strategies
Chemical modifications to mRNA molecules represent a powerful approach to enhance stability, translational efficiency, and immunogenic profile—all critical factors for successful reprogramming. The table below summarizes the primary mRNA modification strategies and their functional impacts:
Table 1: Key mRNA Modification Strategies for Enhanced Reprogramming
| Modification Type | Chemical Approach | Impact on Function | Application in Reprogramming |
|---|---|---|---|
| Nucleoside Substitution | Replacement of uridine with 5-methoxyuridine (5moU) or pseudouridine | Reduces innate immune activation; increases translational efficiency | Enhances protein expression of reprogramming factors without triggering antiviral responses [46] |
| Codon Optimization | Use of synonymous codons to deplete uridine content | Increases mRNA stability and protein yield | Uridine-depleted sequences show improved expression dynamics for reprogramming factors [46] |
| Structural Optimization | Algorithmic design (e.g., LinearDesign) to optimize secondary structure | Improves mRNA half-life and chemical stability | Increases protein expression 2-128 fold compared to conventional codon optimization [47] |
| 5' Capping | Addition of synthetic cap analogs (e.g., CleanCap) | Enhances translational initiation and protects from degradation | Critical for nuclear export and ribosome binding of reprogramming factor mRNAs [46] |
| Poly(A) Tail Engineering | Optimization of tail length (typically 100-150 nucleotides) | Protects against exonucleolytic degradation and enhances translation | Stabilizes reprogramming factor mRNAs during repeated transfection cycles [48] |
Beyond these mRNA modifications, strategic incorporation of microRNA (miRNA) mimics can dramatically enhance reprogramming outcomes. Specifically, the addition of ESC-specific miRNA-367/302s family delivered as mature miRNA mimics synergizes with modified mRNAs to boost reprogramming efficiency. When combined with modified mRNAs, these miRNAs have demonstrated the ability to increase iPSC colony formation from less than 0.5% to over 90% in individually plated primary fibroblasts [27].
Delivery System Optimization
The efficient delivery of mRNA reprogramming components into cells requires careful optimization of both the delivery method itself and the cellular environment during transfection. Different delivery platforms offer distinct advantages for various cell types:
Table 2: Comparison of mRNA Delivery Systems for Reprogramming
| Delivery Method | Mechanism | Advantages | Limitations | Optimal Cell Types |
|---|---|---|---|---|
| Lipid-Based Transfection | Complexation with cationic lipids forming nanoparticles | High efficiency; suitable for high-throughput applications; minimal equipment requirement | Potential cytotoxicity with repeated transfections; variable efficiency across cell types | Fibroblasts, epithelial cells [27] |
| Electroporation | Electrical pulses create temporary pores in cell membrane | Broad applicability; direct cytoplasmic delivery | High cell mortality; requires specialized equipment; difficult for repeated transfections | Immune cells, hematopoietic stem cells [49] |
| Solupore Platform | Physicochemical membrane permeabilization | Superior cell health and survival; suitable for sensitive primary cells | Newer technology with less established protocols | Blood-derived cells (CD34+, T cells, PBMCs) [49] |
| Nucleofection | Combination of electrical parameters and specific solutions | High efficiency in hard-to-transfect cells | Can induce significant stress responses | Primary fibroblasts, iPSCs [25] |
Critical to successful delivery is the optimization of transfection conditions beyond the method itself. Research demonstrates that adjusting the pH of transfection buffers can dramatically impact efficiency. Specifically, increasing the pH of Opti-MEM buffer from the standard 7.3 to 8.2 improved transfection efficiency in primary human fibroblasts from approximately 20% to 65%, as measured by reporter mRNA expression [27]. This optimization was essential for achieving ultra-high reprogramming efficiencies exceeding 90% in low-density cultures.
The timing and frequency of transfections also play crucial roles. A regimen of seven transfections performed at 48-hour intervals provides optimal results, allowing sufficient expression of reprogramming factors while minimizing cytotoxicity [27]. Transfection intervals shorter than 48 hours increase toxicity, while longer intervals (72 hours) significantly reduce reprogramming efficiency due to insufficient factor expression.
Diagram 1: Optimized workflow for high-efficiency mRNA-based reprogramming, incorporating sequential factor delivery and culture condition optimization.
Experimental Protocols and Workflows
Ultra-High Efficiency Reprogramming Protocol for Primary Fibroblasts
This protocol enables reprogramming of up to 90.7% of individually plated human primary neonatal fibroblasts, producing up to 4,019 iPSC colonies from 500 starting cells [27]:
Pre-culture Conditions: Plate human primary fibroblasts at low density (500-1,000 cells per well of a 6-well plate) in knockout serum replacement (KOSR) medium supplemented with bFGF. Use feeder-free culture conditions with extracellular matrix-coated plates.
Transfection Buffer Preparation: Adjust the pH of Opti-MEM transfection buffer to 8.2 using sodium hydroxide. Alternatively, phosphate-buffered saline (PBS) can be used as an effective transfection buffer.
RNA Complex Formation: For each well of a 6-well plate, combine 600 ng of modified mRNA cocktail (encoding OCT4, SOX2, KLF4, cMYC, LIN28, and NANOG) with 20 pmol of miRNA-367/302s mimics in 125 μL of pH-adjusted Opti-MEM. In a separate tube, dilute 7.5 μL of Lipofectamine RNAiMAX in 125 μL of pH-adjusted Opti-MEM. Combine the two solutions and incubate for 10-15 minutes at room temperature.
Transfection Procedure: Add the RNA-lipid complexes dropwise to cells. Perform transfections every 48 hours for a total of seven transfections. Maintain cells between transfections in KOSR medium with daily medium changes.
Colony Expansion: After 14-21 days, manually pick TRA-1-60 positive colonies and transfer to fresh matrix-coated plates. Expand colonies in essential 8 medium or similar defined maintenance medium.
This protocol has been successfully applied to generate clinically relevant, integration-free iPSCs from various human patient fibroblasts under feeder-free conditions, making it suitable for therapeutic applications.
Sequential Factor Delivery for Gene-Edited iPSC Generation
For creating genetically modified iPSCs with knock-in transgenes, a sequential delivery approach significantly enhances efficiency:
Day 0: Donor Template Delivery: Nucleofect 3×10^6 cells with donor DNA plasmid using optimized nucleofection programs (e.g., Lonza 4D Nucleofector with P4 buffer, program CA167). Immediately after nucleofection, recover cells in RPMI medium for 10 minutes before transferring to culture medium.
Day 1: RNP Complex Delivery: Nucleofect cells with preassembled ribonucleoprotein (RNP) complexes containing Alt-R S.p. HiFi Cas9 V3 or Cas12a Ultra and target-specific guide RNA.
Cold Shock Treatment: Incubate cells at 32°C for 24-48 hours post-RNP delivery to enhance homology-directed repair (HDR) efficiency.
Culture and Screening: Return cells to standard culture conditions (37°C) and expand for 7-10 days before screening for successful knock-in events using flow cytometry or PCR-based methods.
This sequential delivery method achieves knock-in efficiencies exceeding 30% in GMP-compliant iPSC lines without requiring antibiotic selection or complex instrumentation, making it ideal for clinical applications [25].
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of optimized mRNA reprogramming requires specific reagents and systems carefully selected for their proven performance:
Table 3: Essential Research Reagents for mRNA Reprogramming
| Reagent Category | Specific Product/System | Function & Application Notes |
|---|---|---|
| Reprogramming mRNAs | 5-methoxyuridine-modified mRNAs (5moU) | Engineered for enhanced stability and reduced immunogenicity; enables high protein expression of reprogramming factors [46] |
| miRNA Enhancers | miRNA-367/302s mimics | Synergistically enhances reprogramming efficiency when combined with modified mRNAs [27] |
| Transfection Reagent | Lipofectamine RNAiMAX | Effective for repeated transfections with minimal cytotoxicity; compatible with pH-optimized buffers [27] |
| Delivery Platform | Solupore system | Non-invasive physicochemical delivery platform ideal for blood-derived cells; maintains high cell viability [49] |
| Culture Matrix | Recombinant vitronectin or laminin-521 | Defined, xeno-free substrates for feeder-free culture supporting iPSC expansion and quality |
| Reprogramming Media | Knockout Serum Replacement (KOSR) medium | Supports reprogramming of low-density fibroblast cultures when combined with optimized transfection [27] |
The optimization of mRNA modifications and delivery systems has dramatically advanced the field of iPSC generation, transforming it from an inefficient process to a robust, clinically applicable technology. The combination of chemically modified mRNAs with optimized delivery protocols enables reprogramming efficiencies previously considered unattainable, while maintaining the integration-free characteristics essential for therapeutic safety.
Future developments will likely focus on further enhancing the precision and specificity of reprogramming through improved mRNA design algorithms like LinearDesign, which optimizes both structural stability and codon usage [47]. Additionally, the application of novel delivery platforms such as the Solupore system will expand the range of accessible somatic cell sources, particularly blood-derived cells that have historically been challenging to reprogram [49]. As these technologies mature, they will pave the way for standardized, GMP-compliant iPSC generation workflows that support the broad clinical translation of iPSC-based therapies across diverse medical applications.
The integration of mRNA-based reprogramming with CRISPR-mediated gene editing represents another promising frontier, enabling the generation of genetically corrected iPSCs from patient-derived somatic cells in a single streamlined process [25]. Such advances will continue to solidify the role of mRNA technology as a cornerstone of regenerative medicine and personalized cell-based therapeutics.
The advent of induced pluripotent stem cells (iPSCs) has revolutionized regenerative medicine, offering unprecedented opportunities for disease modeling, drug discovery, and cell-based therapies. Among various reprogramming methods, mRNA-based reprogramming has emerged as a particularly promising approach for clinical translation due to its non-integrating nature, high efficiency, and defined temporal activity [20]. Unlike early viral vector methods that pose risks of insertional mutagenesis, synthetic mRNA delivery involves transient expression of reprogramming factors, typically the Yamanaka factors (OCT4, SOX2, KLF4, and c-MYC), without genomic integration [11] [3]. This significantly enhances the safety profile of the resulting iPSCs. However, the transition from research-grade iPSCs to clinically applicable products demands rigorous quality control (QC) protocols to ensure genomic stability and purity throughout the entire workflow—from somatic cell reprogramming to differentiated cell products.
This technical guide examines the primary challenges and solutions for ensuring the quality of mRNA-reprogrammed iPSCs intended for clinical applications. We detail specific QC parameters, experimental methodologies, and analytical frameworks essential for validating iPSC lines, with particular emphasis on the unique considerations of mRNA reprogramming platforms. As the field advances toward clinical applications, establishing robust, standardized QC protocols becomes paramount for regulatory approval and patient safety.
Key Challenges in mRNA-Reprogrammed iPSC Quality
While mRNA reprogramming eliminates the risk of genomic integration, it introduces distinct challenges that must be addressed through comprehensive QC strategies.
Table 1: Key Challenges in mRNA-Reprogrammed iPSC Quality Control
| Challenge | Impact on iPSC Quality | QC Consideration |
|---|---|---|
| Transient p53 Activation | Increased risk of apoptosis; reduced reprogramming efficiency [50] | Monitor p53 pathway; consider MDM4 supplementation to suppress p53 transiently [50] |
| Batch-to-Batch Variability | Inconsistent reprogramming efficiency and iPSC quality [51] | Rigorous testing of starting somatic cells and mRNA reagents |
| Epigenetic Memory | Incomplete reprogramming; biased differentiation potential [30] | Epigenomic profiling (DNA methylation, chromatin accessibility) |
| Genetic and Epigenetic Drift During Culture | Genomic instability; acquired mutations [20] [30] | Regular karyotyping and genomic integrity screening throughout passages |
A critical finding from recent studies indicates that epigenetic variation is most strongly associated with genetic variation at the iPSC stage, and this relationship weakens as epigenetic variation increases in differentiated cells [30]. This highlights the importance of controlling for genetic background and carefully monitoring epigenetic changes throughout the differentiation process to ensure consistent and reproducible results.
Essential Quality Control Parameters and Assays
A multi-tiered QC approach is necessary to fully characterize mRNA-reprogrammed iPSCs. The following parameters and corresponding experimental protocols form the foundation of a robust QC program.
Genomic Stability Assessment
Comprehensive genomic assessment ensures the absence of mutations that could compromise safety or function.
Karyotyping and Copy Number Variation (CNV) Analysis:
- Protocol (G-banding Karyotyping): Harvest iPSCs at 70-80% confluence after colcemid treatment (0.1 µg/mL for 45 min). Swell cells in 0.075 M KCl, fix in 3:1 methanol:acetic acid, drop onto slides, and stain with Giemsa. Analyze 20 metaphase spreads for chromosomal abnormalities at passage 5 and every 10 subsequent passages [20].
- Advanced Method (CNV Analysis by SNP Array or NGS): Extract genomic DNA using silica-column based kits. For SNP array, digest 250 ng DNA, label with fluorescent nucleotides, hybridize to array (e.g., Illumina CytoSNP-850K), and analyze with software (e.g. BlueFuse Multi) calling CNVs >100 kb. For whole genome sequencing (WGS), sequence to ~30x coverage and analyze using tools like BWA-MEM and GATK for variant calling [30].
Oncogenic Mutation Screening:
- Protocol (Targeted NGS Panel): Design a custom panel covering 152 genes frequently mutated in cancers (e.g., TP53, PTEN, MYC). Prepare sequencing libraries from 100 ng iPSC DNA, perform hybrid capture, and sequence on Illumina platform (>500x mean coverage). Validate any variants (>5% allele frequency) by Sanger sequencing [20].
Pluripotency and Differentiation Potential Verification
Pluripotency Marker Characterization:
- Protocol (Immunofluorescence): Culture iPSCs on Matrigel-coated plates, fix with 4% PFA, permeabilize with 0.1% Triton X-100, and block with 5% BSA. Incubate with primary antibodies against OCT4, SOX2, NANOG, SSEA-4, and TRA-1-60 (1:200 dilution) overnight at 4°C, then with fluorophore-conjugated secondary antibodies (1:500). Image with confocal microscopy; >90% of cells should express all markers [20].
- Protocol (Flow Cytometry): Dissociate iPSCs to single cells, fix and permeabilize using commercial kits, and stain with the same antibodies as above. Analyze on a flow cytometer; >85% of cells should be positive for pluripotency markers [25].
Trilineage Differentiation Potential:
- Protocol (Embryoid Body Formation): Harvest iPSCs and transfer to low-attachment plates in differentiation medium (DMEM with 20% FBS, 1% NEAA, 0.1 mM β-mercaptoethanol). Culture for 14 days, then analyze germ layer markers: ectoderm (PAX6, NESTIN), mesoderm (Brachyury, SMA), and endoderm (SOX17, FOXA2) via immunostaining or RT-qPCR [20].
Purity and Identity Confirmation
Identity Testing:
- Protocol (Short Tandem Repeat - STR Analysis): Extract genomic DNA from iPSCs and donor somatic cells. Amplify 16 core STR loci (e.g., CSF1PO, D13S317, D16S539, D5S818, D7S820, TH01, TPOX, vWA) using commercial kits. Analyze fragments by capillary electrophoresis and compare profiles between iPSCs and donor cells; all loci must match [20].
Sterility Testing:
- Protocol (Mycoplasma Detection by PCR): Extract nucleic acids from 1 mL spent culture media using silica-membrane columns. Perform PCR with mycoplasma-specific 16S rRNA primers (e.g., MycoSeq kit). Include positive and negative controls; test monthly [20].
Advanced Methodologies for Enhanced QC
GMP-Compliant Gene Editing Verification
Recent advances in gene editing enable the introduction of safety features in iPSCs, such as inducible caspase suicide genes. The following workflow and data illustrate an optimized, GMP-compatible platform for generating such lines.
Table 2: Efficiency of GMP-Compatible iCaspase9 Knock-in and B2M Knock-out
| iPSC Line (Donor) | Knock-in Efficiency | HLA-I Negative Clones (Biallelic Editing) | Validated Clones Post-Screening |
|---|---|---|---|
| R26 (European) | 32% | 18% | 5/24 |
| R36 (US) | 29% | 15% | 4/26 |
Protocol (Serial RNP and Donor Delivery for Knock-in):
- Day -2: Transition cells to richer medium (e.g., mTeSR Plus).
- Day 0: First Nucleofection (Donor Plasmid): Resuspend 3×10^6 iPSCs in P4 nucleofection buffer with donor plasmid (e.g., iCaspase9-pCAG). Use Lonza 4D Nucleofector with program CA-167. Recover cells for 10 min in RPMI medium at room temperature before plating in mTeSR Plus with 10 µM Y-27632.
- Day 1: Second Nucleofection (RNP Complex): Harvest cells, resuspend in P4 buffer with pre-complexed Cas9 RNP targeting the B2M locus. Nucleofect using the same program. Recover in RPMI and plate.
- Day 1-3: Cold Shock: Incubate cells at 32°C for 48 hours.
- Day 7: Screening: Passage cells by limiting dilution into 96-well plates. Expand and screen clones for HLA-I surface expression after IFNγ stimulation (10 ng/mL, 48 h) via flow cytometry to identify biallelically edited (HLA-I negative) clones [25].
Diagram Title: GMP-Compatible Gene Editing Workflow
Epigenetic Landscape and Donor-Specific Variation
DNA methylation profiling and analysis of chromatin accessibility provide critical insights into the epigenetic state of iPSCs.
Protocol (DNA Methylation Analysis by EPIC Array):
- Extract genomic DNA from iPSCs and bisulfite convert 500 ng using commercial kits. Hybridize to Infinium MethylationEPIC BeadChip, scan with iScan system, and process data with R package minfi. Compare to reference methylation signatures of embryonic stem cells; ensure high correlation (R^2 > 0.9) [30].
Protocol (Chromatin Accessibility by ATAC-seq):
- Harvest 50,000 iPSCs, wash in PBS, and lyse with cold lysis buffer. Immediately transpose chromatin with Tr5 transposase (37°C, 30 min). Purify DNA and amplify with indexed primers for 10-12 cycles. Sequence on Illumina platform (minimum 25 million reads). Align to reference genome and call peaks with MACS2; compare to ENCODE ESC datasets [30].
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents for mRNA Reprogramming and QC Workflows
| Reagent / Solution | Function | Example Product / Specification |
|---|---|---|
| Synthetic mRNA Reprogramming Kit | Delivers OSKM factors without genomic integration | StemRNA 3rd Gen Reprogramming Kit [50] |
| MDM4 mRNA | Suppresses p53 activation, enhances PBMC reprogramming efficiency | MDM4-S367A mutant for increased stability [50] |
| iMatrix-511 | Defined, xeno-free substrate for iPSC culture | Recombinant laminin-511 E8 fragment [50] |
| StemFit AK03N | Chemically defined, xeno-free iPSC culture medium | Suitable for reprogramming and maintenance [50] |
| Cas9 HiFi RNP | High-fidelity nuclease for precise gene editing | Alt-R S.p. HiFi Cas9 Nuclease V3 [25] |
| GMP-Compatible Nucleofector | Efficient delivery of nucleic acids and proteins | Lonza 4D Nucleofector System [25] |
Ensuring genomic stability and purity in mRNA-reprogrammed iPSCs requires a multi-faceted QC strategy that addresses the unique challenges of this reprogramming method. By implementing the detailed protocols and frameworks outlined in this guide—from comprehensive genomic integrity checks and pluripotency verification to advanced epigenetic profiling—researchers can establish robust quality control systems that meet the stringent requirements for clinical translation. As mRNA reprogramming technologies continue to evolve, these QC protocols will form the foundation for developing safe and effective iPSC-based therapies.
The field of induced pluripotent stem cell (iPSC) research presents a fundamental paradox: the very oncogenes that enable cellular reprogramming also pose significant safety risks that must be managed for therapeutic applications. The Myc family of transcription factors, particularly c-Myc, serves as a powerful facilitator of somatic cell reprogramming yet constitutes a major barrier to clinical translation due to its well-characterized tumorigenic potential. Within the context of mRNA-based iPSC generation, this balancing act becomes increasingly critical, as transient expression systems seek to harness Myc's reprogramming efficiency while mitigating its oncogenic risks.
The discovery that somatic cells could be reprogrammed to pluripotency using defined factors, including c-Myc, revolutionized regenerative medicine [52] [3]. The original Yamanaka factors (OCT4, SOX2, KLF4, and c-MYC) demonstrated that forced expression of these proteins could epigenetically reset differentiated cells, opening unprecedented opportunities for disease modeling, drug screening, and cell-based therapies [3]. However, the constitutive activation of c-Myc, which is aberrantly expressed in over 70% of human cancers, raised immediate safety concerns [53] [54]. This review examines strategies to balance the reprogramming efficiency afforded by Myc proteins with the development of safer alternatives, with particular emphasis on their application in mRNA-based reprogramming systems.
Myc Biology and Mechanisms in Cellular Reprogramming
Structural and Functional Properties of Myc Proteins
The Myc family consists of three main members in mammalian cells: c-Myc, N-Myc, and L-Myc. These proteins belong to the basic-helix-loop-helix-leucine zipper (bHLHZip) family and function as master transcriptional regulators controlling essential cellular processes including proliferation, metabolism, biosynthesis, and apoptosis [53] [54]. Structurally, Myc proteins contain several conserved domains:
- N-terminal transactivation domain (TAD): Contains conserved Myc boxes (MB0, MBI, MBII) involved in transcriptional regulation and protein degradation
- Central PEST domain: Rich in proline, glutamate, serine, and threonine residues
- C-terminal bHLHZip domain: Responsible for DNA binding and heterodimerization with Max [54]
For transcriptional activation, Myc must heterodimerize with its obligate partner Max. The Myc-Max complex then binds to Enhancer-box (E-box) DNA sequences (CANNTG) in target gene promoters, recruiting histone acetyltransferases and other co-factors to activate transcription [54]. This network regulates approximately 15% of all genes, including those involved in cell cycle progression, ribosome biogenesis, and metabolism [54].
Mechanisms of Myc in Somatic Cell Reprogramming
During iPSC generation, Myc enhances reprogramming efficiency through multiple mechanisms. It promotes the metabolic switch from oxidative phosphorylation to glycolysis, facilitates chromatin remodeling to an open configuration, and accelerates cell cycle progression [52] [3]. Importantly, Myc influences the stochastic phase of reprogramming, increasing the probability that somatic cells overcome barriers to pluripotency.
In the context of mRNA reprogramming, Myc's role becomes particularly nuanced. While traditional viral methods integrate Myc into the host genome, creating persistent expression concerns, mRNA-based delivery offers transient expression that may reduce tumorigenic risks. However, the optimal timing, dosage, and duration of Myc expression remain active areas of investigation.
Table 1: Comparison of Myc Family Members in Cellular Reprogramming
| Property | c-Myc | L-Myc | N-Myc |
|---|---|---|---|
| Reprogramming Efficiency | High | Moderate | High |
| Tumorigenic Potential | High | Lower | High |
| Expression Pattern | Ubiquitous in proliferating cells | Restricted | Restricted |
| Role in Native Development | Embryonic and adult tissues | Specific developmental stages | Neural and renal development |
| Use in mRNA Reprogramming | Common but high risk | Emerging as safer alternative | Limited due to tissue specificity |
Small Molecule Inhibitors as Myc Alternatives
Direct Small Molecule Inhibitors of Myc
The intrinsically disordered nature of Myc proteins has historically made them challenging drug targets, leading to their classification as "undruggable" [53]. However, recent advances have identified several small molecules that directly target Myc:
MYCi975 and MYCi361 represent novel chemotypes identified through in silico screening of large chemical libraries followed by rapid in vivo validation [53]. These compounds bind directly to the MYCHot1 region (amino acids 366-381) within the C-terminal bHLHZip domain, inhibiting MYC/MAX complex formation and promoting MYC degradation via enhanced GSK-3β-mediated phosphorylation at threonine 58 [53] [55].
10074-G5 and 10058-F4 are earlier-generation inhibitors that also disrupt MYC/MAX dimerization through binding to distinct regions of the disordered bHLHZip domain [53] [54]. While these compounds demonstrated proof-of-concept for direct Myc inhibition, their limited potency and poor pharmacokinetic properties restricted their utility as therapeutic agents.
The mechanism of these direct inhibitors involves binding to transiently structured regions within the disordered Myc protein, preventing its interaction with Max and subsequent DNA binding [53]. This inhibition leads to selective downregulation of Myc target genes and impaired proliferation of Myc-dependent cells.
Indirect Myc Targeting Strategies
Alternative approaches to modulating Myc activity include:
- Bromodomain and extraterminal (BET) inhibitors: These compounds target BRD4 proteins, which stabilize Myc transcription, thereby reducing Myc expression [53]
- GSK-3β enhancers: Compounds that enhance GSK-3β-mediated phosphorylation of Myc at T58 promote its proteasomal degradation [53] [54]
- Aurora kinase A inhibitors: Prevent Myc stabilization by interfering with the AURKA-Myc interaction [54]
Table 2: Characteristics of Representative Myc-Targeting Compounds
| Compound | Target | Mechanism | Potency | Therapeutic Index |
|---|---|---|---|---|
| MYCi975 | MYC direct | Binds MYCHot1, promotes degradation | High | Improved |
| 10058-F4 | MYC direct | Binds MYCHot2, disrupts dimerization | Low-moderate | Limited |
| BIX-01294 | Indirect | G9a histone methyltransferase inhibitor | Moderate | N/A |
| VPA | Indirect | HDAC inhibitor | Moderate | N/A |
Experimental Approaches and Methodologies
Assessing Myc Inhibition in Reprogramming Experiments
To evaluate the efficacy of small molecule Myc alternatives in mRNA-based iPSC generation, researchers employ standardized experimental protocols:
Cell Culture and mRNA Transfection:
- Isolate human dermal fibroblasts from tissue biopsies and culture in DMEM supplemented with 10% FBS
- Generate modified mRNA encoding OCT4, SOX2, KLF4, with experimental groups either including or excluding Myc mRNA
- Transfert fibroblasts using lipid-based nanoparticles with 0.5-1μg mRNA per well every 24 hours for 10-18 days
- Supplement experimental groups with small molecule Myc inhibitors at varying concentrations (e.g., MYCi975 at 0.1-10μM)
Reprogramming Efficiency Assessment:
- Monitor emerging iPSC colonies daily, with first colonies typically appearing between days 7-10
- Quantify reprogramming efficiency by counting alkaline phosphatase-positive colonies at day 18
- Confirm pluripotency through immunocytochemistry for NANOG, SSEA4, and TRA-1-60
- Evaluate differentiation potential through embryoid body formation and trilineage differentiation
Safety and Tumorigenicity Analysis:
- Perform karyotype analysis to detect chromosomal abnormalities
- Conduct teratoma assays by injecting 1×10^6 iPSCs into immunodeficient mice
- Monitor tumor formation for 12-20 weeks, with histological examination of resulting tissues
- Evaluate residual Myc expression in differentiated progeny using qRT-PCR and Western blot
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Myc Modulation in iPSC Research
| Reagent Category | Specific Examples | Function in Research |
|---|---|---|
| Reprogramming mRNAs | Modified mRNA encoding OSKM or OSK | Induction of pluripotency without genomic integration |
| Myc Inhibitors | MYCi975, 10058-F4, JQ1 | Evaluate Myc-independent reprogramming and safety |
| Cell Culture Supplements | VPA, sodium butyrate, 2i/LIF | Enhance reprogramming efficiency and stabilize pluripotency |
| Characterization Antibodies | Anti-NANOG, SSEA4, TRA-1-60 | Pluripotency verification |
| Detection Assays | Alkaline phosphatase live stain | Early identification of emerging iPSC colonies |
Visualization of Myc Networks and Experimental Workflows
Myc Signaling and Inhibition Network
mRNA Reprogramming Workflow with Myc Alternatives
The strategic balance between utilizing Myc's reprogramming efficiency and mitigating its oncogenic risks represents a critical frontier in iPSC research. The development of small molecule Myc inhibitors and the optimization of mRNA-based delivery systems together provide a promising path toward generating clinically relevant iPSCs with reduced tumorigenic potential. Current evidence suggests that complete elimination of Myc from reprogramming protocols may be ideal for safety, but comes at the cost of efficiency and kinetics. The future likely lies in precisely timed, dose-controlled Myc expression or inhibition that maximizes reprogramming while minimizing residual oncogene activity.
Emerging technologies such as CRISPR-Cas9 gene editing and AI-guided differentiation are further enhancing our ability to control Myc-related pathways in iPSC derivatives [8] [20]. Additionally, the observation that Myc inhibition modulates the tumor immune microenvironment and upregulates PD-L1 expression suggests potential combination strategies that could address both tumorigenicity and immune rejection [53] [55]. As these technologies mature, the careful balancing of oncogene use will remain central to the safe clinical translation of iPSC-based therapies.
mRNA vs. The Alternatives: A Comparative Analysis of Reprogramming Technologies
The generation of induced pluripotent stem cells (iPSCs) represents a cornerstone of modern regenerative medicine, disease modeling, and drug discovery. Central to this technology is the method of delivering reprogramming factors to somatic cells, a process that significantly influences the safety, efficiency, and applicability of the resulting iPSCs. This whitepaper provides a comprehensive technical comparison of three leading non-integrating reprogramming methods: mRNA-based systems, Sendai virus vectors, and episomal vectors. As the field progresses toward clinical applications, understanding the nuanced advantages and limitations of each platform becomes paramount for researchers and therapy developers. The role of mRNA technology is particularly transformative, offering a non-integrating, controllable, and highly efficient strategy for directing cellular reprogramming [9]. By examining the molecular mechanisms, practical protocols, and performance metrics of these systems, this analysis aims to equip scientists with the data necessary to select the optimal reprogramming methodology for specific research or therapeutic goals.
Technical Comparison of Reprogramming Methods
The choice of reprogramming method involves trade-offs between efficiency, safety, workload, and suitability for different cell types. The following analysis synthesizes data from direct comparative studies and recent technological advances.
Table 1: Key Characteristics of Non-Integrating Reprogramming Methods
| Feature | mRNA Reprogramming | Sendai Virus (SeV) | Episomal Vectors |
|---|---|---|---|
| Molecular Mechanism | Transient transfection of synthetic mRNA encoding reprogramming factors | Transduction with replication-deficient, RNA virus-based vector | Transfection of OriP/EBNA-1 plasmid DNA expressing factors |
| Reprogramming Efficiency | ~2.1% (highest) [56] | ~0.077% [56] | ~0.013% - 0.1% [56] [57] |
| Genomic Integration | No risk (non-integrating) [8] [58] | No risk (cytoplasmic RNA virus) [8] | Low risk; predominantly non-integrating [57] |
| Factor Persistence | Short (days), requires repeated transfections [56] | Medium; cleared over passages (often by passage 10) [56] | Long; slowly lost over passages, can persist [56] |
| Hands-On Workload | High (requires daily transfections) [56] | Low [56] | Medium [56] |
| Time to Colony Emergence | ~14 days (fastest) [56] | ~26 days [56] | ~20 days [56] |
| Aneuploidy Rate | 2.3% (lowest) [56] | 4.6% [56] | 11.5% [56] |
| Ideal Cell Source | Fibroblasts, PBMCs (with optimized protocol) [50] | Fibroblasts, PBMCs [56] | PBMCs, fibroblasts [57] |
| Success Rate | Lower (27%); improved with miRNA (73%) [56] | High (94%) [56] | High (93%) [56] |
| Cost & Biosafety | Costly reagents; minimal biosafety concerns | Expensive virus; requires BSL-2 precautions [57] | Cost-effective; minimal biosafety concerns |
Analysis of Comparative Data
- Efficiency vs. Reliability: While mRNA reprogramming demonstrates the highest theoretical efficiency, its success rate can be sample-dependent and lower than viral and episomal methods without protocol optimizations like co-transfection with microRNAs [56]. Sendai virus and episomal vectors offer high reliability in generating colonies across diverse samples.
- Genetic Safety: All three methods are classified as non-integrating. mRNA is the most definitive in its lack of persistence. Sendai virus is non-integrating but requires monitoring for viral clearance. Episomal vectors are predominantly extrachromosomal but necessitate screening for plasmid retention, as the EBNA1-based plasmids can persist in a small subset of higher-passage lines [56].
- Epigenetic Considerations: A genome-wide DNA methylation profile study comparing iPSCs generated from the same parental somatic cell line revealed that the number of differentially methylated regions (DMRs) relative to embryonic stem cells was often cell-line dependent rather than strictly vector-specific. However, Sendai-virus derived iPSCs consistently showed the lowest number of aberrant methylation sites, while iPSCs generated by non-integrating methods did not show vector-specific DMRs at promoter regions [59].
Detailed Experimental Protocols
mRNA Reprogramming Protocol
The mRNA reprogramming method uses synthetic, modified mRNAs to transiently express reprogramming factors, typically the Yamanaka factors (OCT4, SOX2, KLF4, c-MYC).
Key Reagents:
- StemRNA 3rd Gen Reprogramming Kit (REPROCELL): A commercial kit containing modified mRNAs for the reprogramming factors.
- Transfection Reagent: A carrier to complex with and deliver mRNA into cells.
- iMatrix-511: A recombinant laminin-511 E8 fragment used as a substrate for iPSC culture.
- StemFit AK03N medium: A defined culture medium for iPSC generation and maintenance.
Workflow:
- Day 0 - Seeding Cells: Harvest the somatic cells (e.g., human dermal fibroblasts - HDFs). Mix the cell suspension directly with the synthetic mRNA-transfection reagent complexes and iMatrix-511. Seed this mixture onto a culture plate. The medium used is StemFit AK03N without bFGF [50].
- Days 1-7 - Repeated Transfection: The key to this method is daily transfections for a period (e.g., 4 days minimum for HDFs) to maintain sufficient levels of reprogramming proteins [56] [50].
- Days 7-21 - Colony Formation and Picking: Change to fresh StemFit medium without transfection complexes after the transfection period. Monitor for the emergence of compact, ESC-like colonies. Colonies can be ready for picking as early as day 14 [56].
- Enhancement for Challenging Cells: For difficult-to-reprogram cells like Peripheral Blood Mononuclear Cells (PBMCs), supplementing the reprogramming cocktail with mRNA encoding MDM4 (a p53 suppressor) has been shown to significantly improve efficiency by mitigating stress-induced apoptosis [50].
Sendai Virus Reprogramming Protocol
Sendai virus is an RNA virus that replicates in the cytoplasm without integrating into the host genome, making it a safe and efficient viral delivery method.
Key Reagents:
- Cytotune-iPS Sendai Virus Kit (Thermo Fisher): A common commercial kit containing SeV particles for OCT4, SOX2, KLF4, and c-MYC.
- Feeder Cells or Recombinant Matrix: A layer to support cell growth (e.g., Mitomycin-C-treated mouse embryonic fibroblasts - MEFs).
- Pluripotency Stem Cell Medium: e.g., mTeSR or StemFit.
Workflow:
- Day 0 - Seeding Somatic Cells: Plate the target somatic cells (e.g., fibroblasts) at a high density on feeder cells or a coated surface.
- Day 1 - Viral Transduction: Replace the medium with a fresh medium containing the CytoTune Sendai virus particles. A typical Multiplicity of Infection (MOI) is 3-5 for each virus. Incubate cells with the virus for 12-24 hours.
- Day 2 - Post-Transduction: Remove the virus-containing medium and replace it with fresh somatic cell medium.
- Days 3-26 - Medium Changes and Monitoring: Change the medium every other day. Between days 7-15, a significant morphological change should occur, leading to the appearance of compact iPSC colonies around day 26 [56].
- Picking and Viral Clearance: Pick colonies and expand them. The SeV RNA is gradually diluted and lost with cell passaging. Monitor for the clearance of viral RNA via RT-PCR, as it can persist beyond passage 10 in some lines [56].
Episomal Vector Reprogramming Protocol
This method uses episomal plasmids based on the Epstein-Barr virus OriP/EBNA1 system, which allows for plasmid replication and transient expression of reprogramming factors without genomic integration.
Key Reagents:
- pCXLE Episomal Vectors: A common toolkit includes three plasmids:
pCXLE-hOCT3/4-shp53(Addgene #27077): Encodes OCT3/4 and an shRNA against p53.pCXLE-hSK(Addgene #27078): Encodes SOX2 and KLF4.pCXLE-hUL(Addgene #27080): Encodes L-MYC and LIN28.
- pCXWB-EBNA1 (Addgene #37624): An optional plasmid for transient EBNA1 expression to boost initial efficiency.
- Electroporator (e.g., Neon): For plasmid delivery into cells.
Workflow:
- Day 0 - Electroporation: Isolate and resuspend somatic cells (e.g., PBMCs) in an electroporation buffer with the mixture of episomal plasmids. Deliver the plasmids via electroporation.
- Day 1 - Recovery: Plate the electroporated cells onto a culture dish with recovery medium.
- Days 2-20 - Culture and Monitoring: Change the medium regularly. Colonies typically emerge and are ready for picking around day 20 [56] [57].
- Screening for Plasmid Loss: The episomal plasmids are gradually lost during cell proliferation. However, a fraction of lines may retain them, especially at early passages. It is crucial to screen established iPSC lines for the loss of EBNA1 and other plasmid sequences by PCR [56].
Table 2: Essential Research Reagent Solutions for iPSC Reprogramming
| Reagent Category | Specific Examples | Function in Reprogramming |
|---|---|---|
| Reprogramming Factors | StemRNA 3rd Gen Kit (mRNA), CytoTune-iPS Kit (SeV), pCXLE vectors (Episomal) | Delivery of OSKM or alternative factor combinations to initiate reprogramming. |
| Culture Substrate | iMatrix-511 (Laminin-511), Matrigel, Vitronectin | Provides a defined extracellular matrix for cell adhesion and pluripotency support. |
| Cell Culture Medium | StemFit AK03N, mTeSR1, Pluriton Reprogramming Medium | Nutrient-rich, defined medium optimized for iPSC generation and maintenance. |
| Transfection/Tools | Transfection Reagent (e.g., Lipofectamine), Neon Electroporation System | Enables efficient delivery of nucleic acids (mRNA, DNA) into target somatic cells. |
| Efficiency Enhancers | MDM4/miRNA Booster (for mRNA), shRNA against p53 (for Episomal) | Suppresses innate immune response (mRNA) or senescence pathways to boost colony formation. |
The choice between mRNA, Sendai virus, and episomal vector systems is not a matter of identifying a single superior technology, but rather of selecting the most appropriate tool for a specific application. mRNA reprogramming stands out for its high speed, exceptional efficiency, and definitive non-integration safety profile, making it ideal for projects prioritizing rapid generation of clinical-grade lines from amenable cell sources. Sendai virus vectors offer a robust, reliable, and widely validated platform with high success rates across various cell types, though considerations of cost and viral clearance are necessary. Episomal vectors present a cost-effective and safe non-viral alternative, with particularly strong utility for reprogramming PBMCs, despite a generally lower efficiency that can be mitigated with optimized factor cocktails.
The ongoing evolution of mRNA technology, including improved modifications and delivery systems, solidifies its pivotal role in the future of iPSC research and therapy development [58]. As the field advances toward broader clinical translation, the combination of these reprogramming methods with precise gene-editing tools like CRISPR-Cas9 will further unlock the potential of iPSCs in creating personalized regenerative therapies and sophisticated human disease models [8]. Researchers are thus empowered to make informed, context-driven decisions to accelerate their scientific and therapeutic goals.
The integration of messenger RNA (mRNA) technology with induced pluripotent stem cell (iPSC) research represents a transformative advancement in regenerative medicine and therapeutic development. The reprogramming of somatic cells into pluripotent stem cells using mRNA-based delivery of transcription factors offers a promising alternative to viral vectors, yet introduces distinct safety considerations that require rigorous assessment. This technical guide provides an in-depth analysis of the tumorigenicity and immunogenicity risks associated with mRNA-reprogrammed iPSCs, framing these concerns within the context of their clinical application for researchers, scientists, and drug development professionals. Understanding these safety parameters is crucial for advancing iPSC-based therapies from laboratory research to clinical implementation.
The fundamental safety advantage of mRNA-based reprogramming lies in its non-integrating nature. Unlike viral vectors that permanently incorporate into the host genome, mRNA molecules function episomally in the cytoplasm without genomic integration, significantly reducing the risk of insertional mutagenesis [20] [60]. However, this approach presents unique challenges including transient transgene expression that may necessitate repeated transfections, potential immunostimulatory properties of exogenous mRNA, and the oncogenic potential of reprogramming factors themselves. This whitepaper systematically addresses these challenges through the lens of current scientific evidence and methodological approaches for comprehensive safety profiling.
Tumorigenicity Risks in mRNA-Reprogrammed iPSCs
Oncogenic Potential of Reprogramming Factors
The core reprogramming factors essential for inducing pluripotency present varying degrees of oncogenic risk that must be carefully evaluated in safety assessment protocols. The original Yamanaka factors (OSKM: OCT4, SOX2, KLF4, c-MYC) demonstrate significantly different risk profiles, with c-MYC representing the most substantial concern due to its well-characterized role as a classical oncogene [11] [61]. Research indicates that c-MYC overexpression contributes directly to tumorigenesis, with studies showing it is constitutively and aberrantly expressed in over 70% of human cancers [61]. The oncogenic potential of these factors is not merely theoretical; investigations have revealed that activation of MYC-dependent cancer enhancers can cause genetic mutations in human mammary epithelial cells, establishing a direct mechanistic link between reprogramming factors and oncogenic transformation [61].
Alternative reprogramming factor combinations offer improved safety profiles. The OSNL combination (OCT4, SOX2, NANOG, LIN28) eliminates c-MYC entirely, thereby addressing its associated tumorigenic risks [11]. Similarly, substitution of c-MYC with L-MYC or N-MYC has demonstrated reduced tumorigenic potential while maintaining reprogramming efficiency [11]. Factor-independent approaches using small molecule combinations represent the most promising direction for eliminating oncogene-related tumorigenicity entirely [11] [61]. These chemical reprogramming methods significantly enhance the safety of iPSCs and their potential for clinical applications by completely avoiding the introduction of exogenous oncogenes [11].
Incomplete Reprogramming and Teratoma Formation
A fundamental tumorigenicity concern in mRNA-reprogrammed iPSCs stems from incomplete reprogramming, which can yield partially reprogrammed cells with residual pluripotent characteristics. These cells may evade standard purification and differentiation protocols, potentially leading to teratoma formation following transplantation. The risk is particularly significant with mRNA reprogramming due to the transient nature of factor expression, which may insufficiently maintain the pluripotent state throughout the reprogramming process. Research has demonstrated that grafted iPSC-derived neurospheres can form tumors consisting of Nestin+ undifferentiated neural cells during long-term observation, even when initial transplantation showed functional recovery [62].
The completeness of reprogramming is influenced by multiple technical factors including reprogramming efficiency, which varies considerably between methods. Non-integrating strategies like mRNA reprogramming typically demonstrate lower efficiency (approximately 0.001%) compared to integrating viral approaches [61]. This lower efficiency increases the probability of incomplete reprogramming events. Additionally, studies have identified that late-onset activation of the OCT4 transgene may be associated with tumor formation, highlighting the importance of monitoring transgene persistence and reactivation even in non-integrating approaches [62]. Transcriptome analyses of tumorigenic iPSC derivatives have revealed that a heightened mesenchymal transition may contribute to tumor progression, suggesting epithelial-mesenchymal transition (EMT) pathways as critical monitoring parameters in safety assessment [62].
Genomic and Epigenetic Instability
The reprogramming process itself can induce genomic and epigenetic abnormalities that predispose iPSCs to tumorigenicity. Rapid cellular division during reprogramming can lead to DNA replication stress, resulting in copy number variations (CNVs) and point mutations that may impact tumor suppressor genes or oncogenes. mRNA reprogramming may mitigate some of these risks through its transient nature, but the potential for stress-induced genomic instability remains a concern. Epigenetic memory of the source somatic cells represents another significant consideration, as incomplete epigenetic resetting can persist through reprogramming and influence differentiation potential and tumorigenic propensity [61].
The role of tumor suppressor pathways in maintaining genomic stability during reprogramming is particularly crucial. Suppression of p53, a key tumor suppressor, significantly increases both reprogramming efficiency and tumorigenicity of iPSC-like cells [61]. Studies in mice derived from iPSCs with p53 knockout demonstrated enhanced tumorigenicity, establishing a clear relationship between tumor suppressor pathway manipulation and cancer risk [61]. Therefore, comprehensive genomic and epigenetic profiling represents an essential component of tumorigenicity risk assessment, with particular attention to tumor suppressor pathway integrity and global epigenetic patterning.
Table 1: Strategies to Mitigate Tumorigenicity Risk in mRNA-Reprogrammed iPSCs
| Risk Factor | Specific Concern | Mitigation Strategy | Experimental Assessment |
|---|---|---|---|
| Oncogenic Reprogramming Factors | c-MYC oncogenic activity | Use L-MYC or N-MYC alternatives; chemical reprogramming | Teratoma formation assays; oncogene expression monitoring |
| Incomplete Reprogramming | Residual undifferentiated cells | Optimized differentiation protocols; cell sorting | Flow cytometry for pluripotency markers; long-term in vivo studies |
| Genomic Instability | Accumulation of mutations during reprogramming | Limit culture passages; careful clone selection | Karyotyping; whole-genome sequencing; CNV analysis |
| Epigenetic Abnormalities | Incomplete epigenetic resetting | Epigenetic modifier treatment; extended culture | DNA methylation profiling; histone modification analysis |
Immunogenicity Considerations for mRNA-Derived iPSCs
Innate Immune Recognition of Exogenous mRNA
The immunogenicity profile of mRNA-reprogrammed iPSCs is fundamentally shaped by the innate immune system's capacity to recognize exogenous RNA. Pattern recognition receptors (PRRs), particularly Retinoic Acid Inducible Gene-I (RIG-I) and Toll-like receptors (TLRs), detect molecular patterns associated with viral RNA and can similarly recognize in vitro transcribed (IVT) mRNA used in reprogramming [60]. This recognition triggers type I interferon (IFN) responses and inflammatory cytokine production that may compromise iPSC generation, reduce reprogramming efficiency, and potentially establish a pro-inflammatory microenvironment conducive to immune rejection upon transplantation.
Strategic mRNA engineering approaches have been developed to circumvent these innate immune recognition pathways. Nucleoside modifications represent the most effective strategy, with replacement of uridine with N1-methylpseudouridine (m1Ψ) demonstrating significantly reduced immunogenicity and enhanced translation efficiency compared to unmodified mRNA [60]. This modification was successfully implemented in SARS-CoV-2 mRNA vaccines and has direct applicability to iPSC reprogramming protocols. Additionally, optimization of the 5' cap structure from cap 0 to cap 1 enables evasion of RIG-I detection, while purification methods to remove double-stranded RNA contaminants further minimize innate immune activation [60]. These engineering approaches collectively render therapeutic mRNA less visible to the innate immune system while enhancing its translational capacity and persistence.
Allogene Immune Recognition of Differentiated iPSCs
Beyond innate immune responses to the reprogramming mRNA itself, differentiated iPSC derivatives face potential recognition by the adaptive immune system upon transplantation. Even autologous iPSCs may theoretically elicit immune responses due to epigenetic alterations or antigenic presentation of previously sequestered proteins during differentiation. However, the primary immunogenicity concern for clinical applications involves allogeneic transplantation scenarios, where human leukocyte antigen (HLA) mismatches between donor and recipient can trigger robust T-cell mediated rejection [63]. The polymorphic nature of HLA genes presents significant challenges for matching, with bone marrow registries of millions of donors still failing to find matches for 40-50% of cases for HLA-A and HLA-B loci [63].
Innovative gene editing approaches are being employed to address these allogeneic recognition barriers. Hypoimmunogenic universal iPSCs are being generated through targeted disruption of key HLA genes using CRISPR-Cas9 technology [63]. The most promising strategies involve biallelic knockout of HLA-A and HLA-B combined with monoallelic knockout of HLA-C and disruption of the class II major histocompatibility complex transactivator (CIITA) to eliminate all HLA class II molecules [63]. Additional engineering to introduce immune-modulatory molecules such as HLA-G and CD47 further enhances immune evasion by inhibiting natural killer (NK) cell activation and macrophage phagocytosis, respectively [63]. These comprehensive engineering approaches aim to create "universal donor" iPSC lines that can evade host immune surveillance across broad patient populations.
Table 2: Immunogenicity Risk Mitigation Strategies for mRNA-Reprogrammed iPSCs
| Immunogenicity Source | Underlying Mechanism | Engineering Solution | Validation Method |
|---|---|---|---|
| Innate Immune Activation | RIG-I/TLR recognition of exogenous mRNA | Nucleoside modifications (m1Ψ); cap 1 structure; HPLC purification | IFN expression assays; cytokine profiling |
| T Cell-Mediated Rejection | HLA mismatch in allogeneic transplantation | CRISPR knockout of HLA-A/B/DRA; CIITA disruption | Mixed lymphocyte reaction; T cell proliferation assays |
| NK Cell Activation | Missing self-recognition from HLA elimination | HLA-E or HLA-G knock-in; CD47 overexpression | NK cytotoxicity assays; macrophage phagocytosis tests |
| Autoimmune Reactions | Neoantigen presentation during differentiation | Extensive clone characterization; antigen screening | Immunopeptidome analysis; autoantibody detection |
Methodologies for Safety Profile Assessment
Tumorigenicity Testing Protocols
Comprehensive assessment of tumorigenic potential requires a multi-layered experimental approach encompassing in vitro and in vivo evaluations. The teratoma formation assay represents the gold standard for assessing pluripotency and tumorigenic risk simultaneously. This assay involves injecting test iPSCs into immunodeficient mice (typically NOD-SCID or NSG strains) and monitoring for tumor formation over 12-20 weeks [62]. Histopathological examination of resulting tumors provides critical information about differentiation capacity and the presence of undifferentiated cell populations. For mRNA-reprogrammed iPSCs, long-term observation is particularly crucial, as studies have demonstrated functional recovery can deteriorate over time with concomitant tumor emergence, even when short-term results appear favorable [62].
Advanced molecular characterization provides essential complementary data to in vivo assays. Karyotype analysis identifies gross chromosomal abnormalities, while whole-genome sequencing detects point mutations and copy number variations that may predispose to tumorigenesis [20]. The pluripotency marker expression profile should be thoroughly characterized using flow cytometry and immunocytochemistry for key markers including OCT4, SOX2, NANOG, and SSEA-4 [63]. Particularly for mRNA-reprogrammed iPSCs, specialized attention should be directed to transgene persistence assessment, as residual or reactivated reprogramming factor expression represents a significant tumorigenicity concern. Additionally, functional assays evaluating differentiation capacity across all three germ layers provide critical evidence of complete reprogramming and reduced tumorigenic potential.
Immunogenicity Assessment Frameworks
Rigorous immunogenicity assessment requires comprehensive evaluation of both innate immune activation and adaptive immune recognition. For evaluating responses to the reprogramming mRNA itself, in vitro cytokine profiling following mRNA transfection into antigen-presenting cells provides crucial data on innate immune activation. Measurement of type I interferon (IFN-α/β) and pro-inflammatory cytokines (IL-6, TNF-α) offers quantitative assessment of immunostimulatory potential [60]. The double-stranded RNA content should be rigorously quantified as a critical quality attribute, as dsRNA contaminants represent a potent activator of innate immune pathways.
Assessment of allogeneic immune recognition employs established immunology techniques adapted to iPSC derivatives. The mixed lymphocyte reaction (MLR) serves as a fundamental assay, measuring T-cell proliferation and activation when co-cultured with differentiated iPSC products [63]. For more comprehensive evaluation, immunogenicity testing using central memory T cells and effector memory T cells confirms hypoimmunogenic properties of engineered lines [63]. Following interferon-γ stimulation, HLA expression analysis should be performed to verify successful knockout and assess potential compensatory upregulation of non-targeted HLA molecules [63]. For in vivo validation, humanized mouse models with functional human immune systems provide the most physiologically relevant platform for evaluating immune responses to iPSC-derived transplants prior to clinical trials.
Research Reagent Solutions for Safety Assessment
Table 3: Essential Research Reagents for iPSC Safety Profiling
| Reagent Category | Specific Examples | Research Application | Safety Assessment Role |
|---|---|---|---|
| mRNA Reprogramming Kits | StemRNA NM-RNK; CytoTune-iPS 2.0 Sendai | Non-integrating reprogramming | Reduce insertional mutagenesis risk; improve safety profile |
| CRISPR-Cas9 Systems | Alt-R Sp Cas9 Nuclease; TrueCut Cas9 | HLA gene editing; safety switches | Generate hypoimmunogenic lines; introduce contingency controls |
| Pluripotency Markers | Antibodies against OCT4, SOX2, NANOG, SSEA-4 | Flow cytometry; immunocytochemistry | Assess reprogramming completeness; detect residual undifferentiated cells |
| Differentiation Kits | STEMdiff Trilineage Differentiation Kit | Three-germ layer differentiation | Verify differentiation capacity; assess teratoma risk |
| Cytokine Assays | LEGENDplex; ProcartaPlex arrays | Multiplex cytokine quantification | Evaluate innate immune activation; monitor inflammatory responses |
| Genomic Analysis Tools | KaryoStat; CNV analysis software | Chromosomal abnormality detection | Identify genomic instability; monitor mutational load |
The integration of mRNA technology with iPSC research represents a powerful platform for regenerative medicine, offering solutions to longstanding challenges in gene delivery while introducing distinct safety considerations. Comprehensive assessment of tumorigenicity and immunogenicity risks through the methodologies outlined in this technical guide provides a rigorous framework for evaluating the safety profile of mRNA-reprogrammed iPSCs. The ongoing development of advanced engineering strategies including mRNA modification, CRISPR-based HLA editing, and chemical reprogramming continues to enhance the safety and clinical potential of these transformative technologies. As the field advances toward broader clinical application, maintaining rigorous safety assessment protocols remains paramount for realizing the full therapeutic potential of mRNA-based iPSC technologies while ensuring patient safety.
Visualizations
iPSC Safety Assessment Workflow
HLA Engineering for Immunogenicity Reduction
The success of messenger RNA (mRNA) vaccines during the COVID-19 pandemic marked a significant milestone in therapeutic production, showcasing the platform's potential for rapid development, scalable production, and robust immunogenicity [64]. Unlike traditional vaccine platforms that rely on cultured cells or recombinant proteins, mRNA vaccines are produced through entirely cell-free processes, offering substantial advantages in developmental speed and adaptability to emerging pathogens [64]. This manufacturing paradigm is particularly relevant for induced pluripotent stem cell (iPSC) research, where mRNA technology enables non-integrating gene delivery for reprogramming and differentiation. The core mRNA production process is notably streamlined: once a target sequence is defined, developers design the 5' cap and poly(A) tail, generate a DNA template, and proceed through in vitro transcription (IVT) to yield mRNA drug substance, which is then encapsulated in delivery vehicles such as lipid nanoparticles (LNPs) [65]. This standardized approach, coupled with IVT efficiency that generates four to ten times more mRNA than starting DNA template, creates a compelling case for industrial and clinical production scalability [65].
Comparative Analysis of mRNA Production Systems
Technical Limitations of Conventional Batch Production
Conventional mRNA manufacturing follows a centralized, batch-based model characterized by segmented unit operations executed independently and sequentially [64]. The process typically involves bioreactor-based IVT, enzymatic digestion of DNA templates, multiple filtration steps, and chromatographic purifications, with each stage requiring separate reagents, equipment preparation, cleaning protocols, and quality validation [64]. This inherently discontinuous process architecture introduces significant limitations:
- Operational Inefficiency: Optimal enzymatic activity and mRNA yield occur only within a narrow temporal window during IVT, after which reaction efficiency declines, capping productivity per batch [64].
- Scalability Constraints: Each scale-up effort requires larger bioreactors, increased raw material consumption, and more complex downstream equipment, escalating costs and infrastructure demands [64].
- Quality Variability: Fluctuations in reaction conditions, enzyme activity, or purification efficiency between independent batches pose challenges to consistent product quality [64].
- Limited Responsiveness: Prolonged downtime between batch runs due to cleaning, validation, and equipment changeover leads to suboptimal resource utilization, hindering responsiveness to demand surges [64].
Next-Generation Modular and Continuous-Flow Systems
In response to these challenges, next-generation mRNA manufacturing systems have emerged emphasizing decentralization, modularity, automation, and process intensification [64]. These platforms leverage microfluidics to integrate core steps such as IVT, co-transcriptional capping, and downstream purification into streamlined, automated workflows [64]. Key implementations include:
- BioNTech's BioNTainer: A comprehensive decentralized manufacturing solution comprising International Organization for Standardization-standard shipping containers with interconnected units for mRNA synthesis and LNP formulation. Deployed in Rwanda, this modular system achieved operational status within 8 months (versus 3-5 years for conventional facilities) and reduced production costs by approximately 40% compared to imported vaccines [64].
- Quantoom's Ntensify Platform: A continuous-flow technology using proprietary single-use disposables in modular reactors that scale out in parallel rather than scaling up. Operational data from Afrigen Biologics in South Africa shows the system reduces batch-to-batch variability by 85% and decreases overall production costs by 60% compared to conventional batch manufacturing [64].
- Cytiva's Modular Platforms: Standardized, off-the-shelf functional modules that enhance operational efficiency while ensuring compliance with Good Manufacturing Practice (GMP). These systems allow customization of production lines without extensive infrastructure modifications, enabling pharmaceutical companies to quickly adapt manufacturing capabilities to specific therapeutic demands [66].
The table below provides a quantitative comparison of these next-generation platforms against conventional batch production:
Table 1: Performance Comparison of mRNA Manufacturing Platforms
| Performance Metric | Conventional Batch | BioNTainer | Quantoom Ntensify |
|---|---|---|---|
| Deployment Time | 3-5 years | ~8 months | 3 months (optimization) |
| Production Cost Reduction | Baseline | ~40% | ~60% |
| Batch Consistency | High variability | Not specified | 85% improvement |
| Annual Output Capacity | Facility-dependent | Up to 50 million doses | ~3 million doses per reactor run |
| Key Challenges | Scalability limitations, high capital cost | Regulatory harmonization, material dependency | Single-use waste, technical complexity |
mRNA Production Workflows: From Synthesis to Formulation
Core Manufacturing Process
The foundational mRNA production process follows a standardized workflow that enables platform-based manufacturing across different therapeutic applications:
- DNA Template Preparation: Plasmid DNA is engineered to contain the target sequence flanked by regulatory elements, including a bacteriophage promoter (e.g., T7, SP6), 5' and 3' untranslated regions (UTRs), and a poly(A) tail sequence [65].
- In Vitro Transcription (IVT): The DNA template is mixed with recombinant RNA polymerase, nucleotide triphosphates (NTPs), capping reagents, and buffer components to catalyze mRNA synthesis in a cell-free system [64].
- Downstream Processing: The crude IVT reaction mixture undergoes purification through enzymatic digestion of the DNA template, filtration steps to remove impurities, and chromatographic purification (ion exchange, hydrophobic interaction, size exclusion) [64].
- Lipid Nanoparticle (LNP) Formulation: Purified mRNA is encapsulated in LNPs via microfluidic mixing, combining mRNA with ionizable lipids, phospholipids, cholesterol, and PEG-lipids in precise ratios [64] [65].
- Purification and Buffer Exchange: Formulated mRNA-LNPs undergo tangential flow filtration to remove residual solvents and exchange buffers into final formulation buffers [64].
- Quality Control and Fill-Finish: Products undergo comprehensive analytical testing (identity, potency, purity) before aseptic filling into vials or syringes [64].
Process Intensification Through Continuous Manufacturing
Next-generation systems enhance this workflow through process intensification and continuous manufacturing. The diagram below illustrates a streamlined continuous-flow mRNA production system:
This continuous approach maintains sustained enzymatic activity during IVT, enables reduced byproduct accumulation through immediate purification, and incorporates automated process control for real-time quality monitoring [64]. These systems demonstrate superior performance across multiple metrics, as shown in the comparative analysis below:
Table 2: Efficiency Metrics of Batch vs. Continuous mRNA Production
| Process Parameter | Batch Production | Continuous Production |
|---|---|---|
| Productivity & Yield | Lower | Higher |
| Production Consistency | Variable | Highly consistent |
| Reagent Utilization | Decreases over reaction | Sustained efficiency |
| Cost Efficiency | Lower | Higher |
| Scalability | Limited | Highly scalable |
| Byproduct Accumulation | Increased over time | Sustained low levels |
| System Complexity | Lower | Higher initial complexity |
mRNA Applications in iPSC Research and Manufacturing
Enabling Non-Integrating Reprogramming
In iPSC research, mRNA technology has revolutionized somatic cell reprogramming by enabling non-integrating gene delivery that eliminates risks associated with viral vector integration. The standard approach involves:
- Reprogramming Factor Design: mRNA sequences are engineered to encode key pluripotency factors (OCT4, SOX2, KLF4, c-MYC) with modified nucleosides to reduce innate immune recognition [8].
- Sequential mRNA Delivery: Synthetic mRNAs are transfected into somatic cells (e.g., fibroblasts) using non-viral methods, with repeated transfections over 2-3 weeks to maintain reprogramming factor expression [8].
- Reprogramming Trajectory: Exogenous mRNA expression initiates epigenetic remodeling, including DNA demethylation at pluripotency promoter regions and histone modification, driving cells toward pluripotency through partially characterized intermediate states [3] [19].
- iPSC Characterization: Emerging colonies are isolated and evaluated for pluripotency marker expression (NANOG, SSEA-3/4), differentiation potential, and genomic integrity [19].
This mRNA-based reprogramming approach offers significant advantages for clinical-grade iPSC generation, including elimination of genomic integration, precise control over factor expression, and compatibility with GMP manufacturing requirements [8] [19].
CRISPR-Based Genome Editing in iPSCs
Beyond reprogramming, mRNA serves as an optimal delivery format for CRISPR-based genome editing in iPSCs, as demonstrated in recent advances:
- Efficient Knock-in Workflows: A 2025 study established a GMP-compatible, virus-free knock-in method using sequential delivery of Cas9/Cas12a ribonucleoprotein (RNP) complexes and donor DNA templates, achieving >30% knock-in efficiency without antibiotic selection [25].
- Universal Donor iPSC Lines: Researchers created homozygous iPSC lines depleted of HLA class I and carrying an inducible caspase-9 suicide gene, demonstrating the potential for immune-compatible allogeneic therapies [25].
- Epigenome Editing Applications: Japanese researchers employed CRISPR-based epigenome editing to demethylate the Prader-Willi syndrome imprinting control region in patient-derived iPSCs, successfully reactivating silenced maternal genes [67].
The table below outlines key research reagents essential for implementing these mRNA-based iPSC applications:
Table 3: Essential Research Reagents for mRNA-Based iPSC Applications
| Reagent Category | Specific Examples | Function in iPSC Workflow |
|---|---|---|
| Reprogramming mRNAs | Modified mRNA encoding OCT4, SOX2, KLF4, c-MYC | Induction of pluripotency in somatic cells without genomic integration |
| Genome Editing Enzymes | Cas9 mRNA, Cas12a mRNA | CRISPR-mediated gene knockout or knock-in when complexed with guide RNAs |
| Nucleotide Modifications | N1-methylpseudouridine, 5-methylcytidine | Reducing innate immune recognition of synthetic mRNA |
| Lipid Nanoparticles | Ionizable lipids, PEG-lipids, cholesterol | Protecting and delivering mRNA to cells via encapsulation |
| DNA Template Systems | PCR-amplified templates, plasmid DNA | Serving as templates for in vitro transcription of mRNA |
Industrial Scalability and Economic Considerations
CDMO Landscape and Market Dynamics
The growing complexity of mRNA therapeutics has driven increased reliance on Contract Development and Manufacturing Organizations (CDMOs) specializing in mRNA production. The global mRNA therapeutics CDMO market was estimated at $5.0 billion in 2024 and is projected to reach $9.0 billion by 2030, growing at a compound annual growth rate of 10.3% [68]. This growth is fueled by several factors:
- Pipeline Expansion: Over 70 mRNA candidates have entered clinical trials spanning infectious disease vaccines, cancer immunotherapies, protein replacement therapies, and cell/gene editing applications [65].
- Capital Efficiency: CDMOs reduce infrastructure investment for biotech companies, with specialized providers offering end-to-end services from process development to commercial manufacturing [68].
- Technology Access: CDMOs provide access to proprietary manufacturing technologies, including continuous production systems, LNP formulation expertise, and analytical method development [68].
Major players in the mRNA CDMO space include Lonza Group, Thermo Fisher Scientific, Catalent, Inc., and Curia Global, Inc., with increasing competition driving innovation in manufacturing efficiency and cost reduction [68].
Regional Manufacturing Initiatives
Recent years have witnessed significant expansion of regional manufacturing capabilities to enhance supply chain resilience and promote equitable vaccine access:
- Moderna's Global Network: Moderna is bringing three new manufacturing facilities online in 2025 located in the UK (100 million dose capacity), Australia (100 million doses), and Canada (30-100 million doses) to bolster pandemic readiness [69].
- mRNA Technology Transfer Program: Initiatives such as the WHO's mRNA Technology Transfer Program aim to establish sustainable vaccine manufacturing ecosystems in low- and middle-income countries, enhancing global health equity [66].
- African Manufacturing Hubs: The successful deployment of BioNTech's BioNTainer in Rwanda and Quantoom's Ntensify platform at Afrigen Biologics in South Africa demonstrates the feasibility of decentralized mRNA manufacturing in resource-limited settings [64].
Future Directions and Manufacturing Innovation
Emerging Technology Platforms
Next-generation mRNA manufacturing is evolving toward increasingly automated, closed-system platforms that enhance efficiency while reducing contamination risks and operational costs [68]. Key innovations include:
- Self-Amplifying RNA (saRNA): Replicon-based RNA vectors that enable longer-lasting protein expression from lower doses, reducing production demands for dose-intensive applications [64].
- Circular RNA (circRNA): Engineered circular RNA constructs with superior stability that enable prolonged therapeutic protein expression from single administrations [68].
- AI-Optimized Manufacturing: Artificial intelligence and machine learning applications for optimizing mRNA sequence design, predicting secondary structures, and refining production parameters to increase yield and reduce waste [68].
- Thermostable Formulations: Advanced LNP compositions and lyophilization approaches that reduce or eliminate cold-chain requirements, enhancing distribution efficiency in resource-limited settings [64].
Advanced Applications in Cell Therapy Manufacturing
The convergence of mRNA and iPSC technologies is creating new paradigms for scalable cell therapy manufacturing:
- In Vivo Cell Reprogramming: mRNA-LNP systems designed for direct in vivo delivery of reprogramming factors to convert resident cells into therapeutic cell types, bypassing ex vivo manipulation [65].
- Universal Off-the-Shelf Therapies: mRNA-edited iPSC lines with knocked-out HLA genes and incorporated safety switches (e.g., inducible caspase-9) that enable allogeneic cell therapies without immune rejection [25].
- Direct Lineage Conversion: mRNA-based transcription factor delivery for direct conversion between somatic cell types without transitioning through pluripotency, potentially accelerating regenerative medicine applications [3].
The implementation of these advanced applications will require further refinement of mRNA manufacturing platforms to ensure consistent quality, scalability, and cost-effectiveness as therapeutic paradigms evolve.
mRNA technology represents a transformative approach to biotherapeutic production that offers distinct advantages in efficiency, scalability, and versatility compared to traditional biological manufacturing platforms. The inherent simplicity of the mRNA production process, combined with emerging continuous manufacturing systems, enables rapid, cost-effective production of diverse therapeutic modalities. For the iPSC field, mRNA platforms provide essential tools for non-integrating reprogramming, precise genome editing, and directed differentiation that are compatible with clinical-grade manufacturing requirements. As the therapeutic landscape evolves toward increasingly personalized and cell-based medicines, the synergy between mRNA production technology and iPSC research will play a pivotal role in enabling scalable, economically viable manufacturing paradigms for next-generation therapies.
The advent of induced pluripotent stem cell (iPSC) technology has revolutionized regenerative medicine and disease modeling. Among the various reprogramming methods, mRNA-based reprogramming has emerged as a superior non-integrating approach, offering an exceptional safety profile, high efficiency, and clinical relevance. This technical review provides a comprehensive analysis of mRNA reprogramming, detailing its molecular mechanisms, optimized protocols, and direct quantitative comparisons with alternative technologies. We position synthetic mRNA as the leading non-integrating method for iPSC generation, providing researchers with actionable experimental frameworks and resource guidelines for implementation.
The foundation of iPSC technology was established by Takahashi and Yamanaka's landmark discovery that somatic cells could be reprogrammed into pluripotent stem cells using defined transcription factors [3]. The original OSKM factors (OCT4, SOX2, KLF4, and c-MYC) delivered via integrating retroviral vectors demonstrated proof-of-concept but raised significant safety concerns regarding genomic integration and tumorigenicity [11] [70]. This prompted the development of non-integrating methods including mRNA reprogramming, episomal plasmids, Sendai virus vectors, and self-replicating RNAs [20].
Non-integrating methods eliminate the risk of insertional mutagenesis but vary significantly in efficiency, reliability, and clinical applicability. Among these, mRNA reprogramming has demonstrated distinct advantages in safety control, reproducibility, and scalability [71]. The transient nature of mRNA molecules in the cellular environment ensures that reprogramming factors are expressed without genetic modification, making this approach particularly suitable for clinical-grade iPSC generation [70].
Molecular Mechanism of mRNA Reprogramming
Mechanistic Basis
mRNA reprogramming operates through the direct introduction of in vitro transcribed (IVT) mRNA molecules encoding reprogramming factors into the cytoplasm of somatic cells. Unlike DNA-based methods, mRNA transcripts bypass the nuclear barrier and are immediately translated into functional proteins by the host cell's ribosomes [71]. This direct translation mechanism enables rapid onset of reprogramming factor expression within hours of transfection.
The reprogramming process initiates with the silencing of somatic genes and activation of early pluripotency networks, progressing through distinct molecular phases [3]. Key events include mesenchymal-to-epithelial transition (MET), epigenetic remodeling through demethylation of pluripotency promoters, and metabolic shift from oxidative phosphorylation to glycolysis [3]. The transient nature of mRNA (typically degraded within 24-48 hours) necessitates repeated transfections to maintain sufficient reprogramming factor levels throughout this multi-phase process [71].
Bypassing Innate Immune Recognition
A critical challenge in mRNA reprogramming is the inherent immunogenicity of exogenous RNA, which triggers pattern recognition receptors and activates antiviral interferon responses [71]. This obstacle has been overcome through two primary strategies:
- Nucleoside Modification: Incorporation of modified nucleosides such as pseudouridine (Ψ) and 5-methylcytidine replaces uridine and cytidine, effectively evading detection by Toll-like receptors [71].
- Interferon Suppression: Supplementation with interferon pathway inhibitors such as B18R protein, which binds and neutralizes type I interferons, significantly enhances cell viability and reprogramming efficiency [71].
The diagram below illustrates the optimized workflow for mRNA reprogramming, incorporating these key modifications:
Comparative Analysis of Non-Integrating Reprogramming Methods
The table below provides a systematic comparison of the major non-integrating iPSC reprogramming methods, highlighting the superior positioning of mRNA-based approaches:
Table 1: Quantitative and Qualitative Comparison of Non-Integrating iPSC Reprogramming Methods
| Parameter | mRNA Reprogramming | Self-Replicating RNA | Sendai Virus | Episomal Vectors |
|---|---|---|---|---|
| Reprogramming Efficiency | 0.2-1.5% [71] | ~1.4% (up to 2-fold higher than mRNA) [71] | 0.1-1% [70] | ~0.01% (significantly lower) [70] |
| Time to iPSC Colonies | 3-4 weeks [71] | 3-4 weeks [71] | 3-4 weeks [70] | 4-5 weeks [70] |
| Genomic Integration Risk | None [71] [70] | None (no DNA intermediate) [71] | Low (persists for ~10 passages) [70] | Low (cleared in ~3 weeks) [70] |
| Transgene Persistence | None (degraded within 24h) [71] | 1-2 weeks (limited persistence) [71] | Up to 10 passages [70] | ~17-21 days [70] |
| Ease of Use | Moderate (requires daily transfections) [71] | Simple (single transfection) [71] | Simple (single infection) [70] | Simple (single transfection) [70] |
| Key Advantages | Footprint-free, well-defined components, clinical suitability [71] [70] | Single transfection, extended protein expression [71] | High efficiency for difficult cells [70] | Simple, cost-effective [70] |
| Key Limitations | Daily transfections, interferon response [71] | Requires B18R, viral sequence presence [71] | Lengthy clearance verification [70] | Very low efficiency, requires oncogenes [70] |
The data demonstrates that while self-replicating RNA offers marginally higher efficiency through reduced transfrequency frequency, mRNA reprogramming provides the most definitive safety profile with no persistence concerns. Sendai virus methods, while popular, require extensive passaging to confirm clearance, complicating their use in clinical applications [70]. Episomal vectors suffer from critically low efficiency without additional oncogenic factors [70].
Optimized mRNA Reprogramming Protocol
Critical Reagent Formulations
Successful mRNA reprogramming requires carefully optimized reagents and conditions. The table below details essential research reagent solutions:
Table 2: Essential Research Reagent Solutions for mRNA Reprogramming
| Reagent/Condition | Function | Optimization Notes |
|---|---|---|
| Modified mRNA Cocktail | Encodes reprogramming factors (OSKM ± Lin28) with enhanced stability and reduced immunogenicity | Incorporate pseudouridine and 5-methylcytidine; cap structure essential for translation [71] |
| B18R Protein | Binds and neutralizes type I interferons to suppress innate immune response | Crucial for cell viability; typically included throughout reprogramming phase [71] |
| Transfection Reagent | Facilitates cellular uptake of mRNA molecules | Polymer- or lipid-based systems optimized for minimal cytotoxicity [71] |
| Basal Medium | Supports both somatic cells and emerging iPSCs | Often DMEM/F12 with specific supplements [72] |
| Small Molecules | Enhances reprogramming efficiency | VPA, sodium butyrate, or 8-Br-cAMP can improve efficiency 2-6 fold [11] |
| Matrix Substrate | Provides structural support for colony formation | Matrigel, vitronectin, or laminin-521 [72] |
Step-by-Step Workflow
The following comprehensive protocol has been optimized for high-efficiency iPSC generation from human fibroblasts:
Day -2: Cell Plating
- Plate 50,000-100,000 human fibroblasts per well of a 6-well plate in standard fibroblast medium
- Ensure cells are 70-90% confluent at time of first transfection
Days 1-17: Daily mRNA Transfection
- Prepare transfection complex: Combine 1.2μg total mRNA (OSKM factors ± Lin28) with appropriate transfection reagent in serum-free medium [71]
- Incubate complex for 15-20 minutes at room temperature
- Replace cell culture medium with fresh medium containing B18R (0.5μg/mL)
- Add transfection complex dropwise to cells
- Repeat transfection daily for 16-17 days with complete medium change
Days 5-21: Medium Transition
- Gradually transition to iPSC culture medium (e.g., mTeSR or E8) as colonies begin to emerge
- Continue daily medium changes with B18R supplementation until day 21
Days 21-28: Colony Selection and Expansion
- Identify and mechanically pick well-defined iPSC colonies with sharp borders
- Transfer to feeder-free culture plates pre-coated with appropriate matrix
- Expand clonal lines for characterization and banking
The strategic interplay of these components is visualized below:
Applications and Clinical Translation
mRNA-reprogrammed iPSCs have become preferred for clinically-oriented applications due to their completely footprint-free nature. These cells are being utilized across multiple domains:
Disease Modeling
iPSCs generated via mRNA reprogramming provide pristine platforms for disease modeling, particularly for neurological disorders like amyotrophic lateral sclerosis (ALS) [11]. Patient-specific iPSC-derived motor neurons enable investigation of disease-specific pathology and molecular mechanisms without confounding genetic integration artifacts [11] [72].
Drug Discovery and Toxicity Testing
Pharmaceutical companies increasingly employ mRNA-iPSCs for toxicity testing and drug screening, offering accurate human-based alternatives to animal models [73]. The defined nature of mRNA reprogramming ensures consistent results across batches, essential for high-throughput screening applications.
Cell Therapy Development
Multiple companies are advancing mRNA-iPSC-derived therapies toward clinical applications. Bayer's subsidiary BlueRock Therapeutics, Fujifilm Cellular Dynamics, and Aspen Neuroscience are among those developing iPSC-derived therapies for Parkinson's disease, heart failure, and ocular diseases [34]. The absence of genomic integration facilitates regulatory approval by eliminating insertional mutagenesis concerns.
Based on comprehensive analysis of efficiency, safety, and practical implementation factors, mRNA reprogramming rightfully claims its position as the leading non-integrating method for iPSC generation. While alternative methods like self-replicating RNA offer marginal efficiency advantages and Sendai virus provides broad cell type compatibility, mRNA technology delivers the optimal balance of clinical safety, experimental control, and reproducibility. The completely footprint-free nature of mRNA-reprogrammed iPSCs, coupled with well-defined reagents and increasingly streamlined protocols, establishes this methodology as the gold standard for applications requiring the highest safety standards, particularly clinical translation and pharmacologic screening. As the field advances toward broader therapeutic application, mRNA reprogramming provides the foundational technology for generating clinically-compliant iPSCs with minimal safety concerns.
Conclusion
mRNA technology has unequivocally established itself as a cornerstone of modern iPSC generation, offering an unambiguously 'footprint-free,' efficient, and clinically promising reprogramming methodology. Its transient nature eliminates the risk of genomic integration, directly addressing the critical safety concerns of oncogenicity associated with earlier viral methods. While challenges such as managing innate immune responses and further optimizing protocols remain, the trajectory of mRNA reprogramming points toward a future of industrialized, clinical-grade iPSC production. Continued investment in refining delivery systems, enhancing reprogramming efficiency with small molecules, and developing robust differentiation protocols will be paramount. The convergence of mRNA reprogramming with advanced gene editing tools like CRISPR-Cas9 and sophisticated 3D organoid models paves the way for a new era in personalized regenerative medicine, disease modeling, and drug discovery, ultimately unlocking the full therapeutic potential of patient-specific stem cells.
References
- 1. Induced pluripotent stem cell [https://en.wikipedia.org/wiki/Induced_pluripotent_stem_cell]
- 2. Induction of pluripotent stem cells from mouse embryonic ... [https://pubmed.ncbi.nlm.nih.gov/16904174/]
- 3. Induced pluripotent stem cells (iPSCs) [https://www.nature.com/articles/s41392-024-01809-0]
- 4. Molecular mechanisms of induced pluripotency - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4322534/]
- 5. Induced Pluripotent Stem Cell (iPSC) Industry Trends for ... [https://bioinformant.com/ipsc-industry-trends/]
- 6. Yamanaka factors critically regulate the developmental ... [https://www.nature.com/articles/cr2008309]
- 7. The emerging promise of induced pluripotent stem cells in ... [https://www.sciencedirect.com/science/article/abs/pii/S1465324925008461]
- 8. Pluripotent Stem Cells: Recent Advances and Emerging ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12025069/]
- 9. mRNA-based technology for engineered regenerative ... [https://www.sciencedirect.com/science/article/pii/S305056232500176X]
- 10. mRNA-Based Genetic Reprogramming - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6453511/]
- 11. Advances in Induced Pluripotent Stem Cell Reprogramming ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12577567/]
- 12. Vectorology and Factor Delivery in Induced Pluripotent Stem Cell Reprogramming - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4046209/]
- 13. Application of Modified mRNA in Somatic Reprogramming to Pluripotency and Directed Conversion of Cell Fate - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8348611/]
- 14. Transient non-integrative expression of nuclear ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7093390/]
- 15. iPSC-based Platforms Market 2025 Report and Trends [https://www.towardshealthcare.com/insights/ipsc-based-platforms-market-sizing]
- 16. Highly efficient reprogramming to pluripotency and directed... [https://pubmed.ncbi.nlm.nih.gov/20888316/]
- 17. HSCI researchers achieve major breakthrough in cell ... [https://www.hsci.harvard.edu/hsci-researchers-achieve-major-breakthrough-cell-reprogramming]
- 18. High-efficiency RNA -based reprogramming ... | Nature Communications [https://www.nature.com/articles/s41467-018-03190-3?error=cookies_not_supported&code=7086c27c-324f-46f3-bca6-1ec71f2692b7]
- 19. A comprehensive analysis of induced pluripotent stem cell ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1593207/full]
- 20. Clinical translation of human iPSC technologies: advances ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1627149/full]
- 21. Research progress of mRNA vaccines for infectious diseases | European Journal of Medical Research | Full Text [https://eurjmedres.biomedcentral.com/articles/10.1186/s40001-025-03060-x]
- 22. Application of induced pluripotent stem cells in the conservation of endangered animals | Stem Cell Research & Therapy | Full Text [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-025-04392-5]
- 23. iPSC Reprogramming Cocktail [https://www.ubrigene.com/ipsc-products/ipsc-reprogramming-cocktail/]
- 24. Generation and purification of iPSC-derived cardiomyocytes ... [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-025-04319-0]
- 25. Sequential factor delivery enables efficient workflow for ... [https://www.nature.com/articles/s41598-025-17876-4]
- 26. mRNA-Based Genetic Reprogramming - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6453511/]
- 27. High-efficiency RNA-based reprogramming of human ... [https://www.nature.com/articles/s41467-018-03190-3]
- 28. Generation of human induced pluripotent stem cells using ... [https://www.sciencedirect.com/science/article/pii/S187350611630006X]
- 29. Multifaceted regulation of somatic by cell ... reprogramming mRNA [https://pubmed.ncbi.nlm.nih.gov/24630793/]
- 30. iPSCs and iPSC-derived cells as a model of human ... [https://www.nature.com/articles/s41467-025-56569-4]
- 31. Successful Delivery of Small Non-Coding RNA Molecules ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12561723/]
- 32. Engineered iPSC‐derived natural killer cells [https://pmc.ncbi.nlm.nih.gov/articles/PMC12246386/]
- 33. Modeling rare genetic disease with patient-derived ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12317316/]
- 34. The Pipeline for of iPSC-Derived Cell Therapeutics in 2026 [https://bioinformant.com/ipsc-derived-cell-therapeutics/]
- 35. ESMO 2025: mRNA-based COVID vaccines generate ... [https://www.mdanderson.org/newsroom/research-newsroom/-esmo-2025--mrna-based-covid-vaccines-generate-improved-response.h00-159780390.html]
- 36. Clinical translation of human iPSC technologies [https://pmc.ncbi.nlm.nih.gov/articles/PMC12498231/]
- 37. Recent progress in the use of induced pluripotent stem ... [https://link.springer.com/article/10.1007/s44340-025-00042-x]
- 38. Induced pluripotent stem cells carrying novel APTX ... [https://www.nature.com/articles/s41420-025-02723-2]
- 39. Large-scale drug screening in iPSC-derived motor neurons ... [https://www.nature.com/articles/s41593-025-02118-7]
- 40. Editorial: Role of induced pluripotent stem cells (iPSCs) in ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2025.1734717/full]
- 41. Type I interferon response impairs differentiation potential ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6347712/]
- 42. The mRNA component of LNP-mRNA vaccines triggers ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1670350/full]
- 43. The mRNA component of LNP-mRNA vaccines triggers ... [https://pubmed.ncbi.nlm.nih.gov/41169401/]
- 44. Identification of Potential Therapeutic Agents for Type I ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12484275/]
- 45. The Generation of iPSCs Expressing Interferon-Beta Under ... [https://www.mdpi.com/1422-0067/26/17/8376]
- 46. Chemical modifications of adenine base editor mRNA and ... [https://www.nature.com/articles/s41467-020-15892-8]
- 47. Algorithm for optimized mRNA design improves stability and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10499610/]
- 48. mRNA-LNP vaccines: rational design, delivery optimization ... [https://pubmed.ncbi.nlm.nih.gov/41251047/]
- 49. Efficient Non-Invasive Delivery of RNA for Somatic ... [https://www.sciencedirect.com/science/article/abs/pii/S1465324925005791]
- 50. MDM4 enables efficient human iPS cell generation from ... [https://www.nature.com/articles/s41598-025-16446-y]
- 51. Engineering the next generation of allogeneic CAR cells [https://pmc.ncbi.nlm.nih.gov/articles/PMC12277817/]
- 52. Induced pluripotent stem cells: applications in regenerative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4313779/]
- 53. Turning up the Heat on MYC: progress in small molecule ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7855142/]
- 54. Taking the Myc out of cancer: toward therapeutic strategies to ... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-020-01291-6]
- 55. Small-Molecule MYC Inhibitors Suppress Tumor Growth ... [https://www.sciencedirect.com/science/article/pii/S1535610819304398]
- 56. A comparison of non-integrating reprogramming methods [https://pmc.ncbi.nlm.nih.gov/articles/PMC4329913/]
- 57. pCXLE toolkit: Efficient episomal plasmid-based method to ... [https://blog.addgene.org/pcxle-toolkit-efficient-nonviral-method-to-reprogram-peripheral-blood-cells-to-ipscs]
- 58. Current landscape of mRNA technologies and delivery ... [https://jbiomedsci.biomedcentral.com/articles/10.1186/s12929-024-01080-z]
- 59. Epigenetic-scale comparison of human iPSCs generated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6222281/]
- 60. mRNA-based vaccines and therapeutics: an in-depth ... [https://jbiomedsci.biomedcentral.com/articles/10.1186/s12929-023-00977-5]
- 61. Possible Strategies to Reduce the Tumorigenic Risk of ... [https://www.mdpi.com/1422-0067/25/10/5177]
- 62. Long-Term Safety Issues of iPSC-Based Cell Therapy in a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4375796/]
- 63. Generation of hypoimmunogenic universal iPS cells ... [https://www.nature.com/articles/s12276-025-01422-3]
- 64. Advancements and challenges in next-generation mRNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12599430/]
- 65. mRNA Therapeutics Are Just Getting Started [https://www.pharmasalmanac.com/articles/mrna-therapeutics-are-just-getting-started-charting-a-post-pandemic-path-forward]
- 66. Modular manufacturing: The key to unlocking mRNA ... [https://www.clinicaltrialsarena.com/sponsored/modular-manufacturing-the-key-to-unlocking-mrna-expansion/]
- 67. CMN Weekly (31 October 2025) [https://crisprmedicinenews.com/news/cmn-weekly-31-october-2025-your-weekly-crispr-medicine-news/]
- 68. mRNA Therapeutics Contract Development & ... [https://www.businesswire.com/news/home/20250903654820/en/mRNA-Therapeutics-Contract-Development-Manufacturing-Organization-Business-Research-2025-2030-Growing-Shift-Towards-Modular-and-Automated-Manufacturing-Solutions-Promotes-Growth-and-Efficiency---ResearchAndMarkets.com]
- 69. Moderna eyes mRNA manufacturing efficiencies as 3 ... [https://www.bioprocessintl.com/facilities-capacity/moderna-eyes-mrna-manufacturing-efficiencies-as-3-plants-set-to-come-online-in-2025]
- 70. The Challenges to Advancing Induced Pluripotent Stem ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10768945/]
- 71. Generation of iPSCs by Nonintegrative RNA-Based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6607707/]
- 72. Quantitative comparison of the mRNA content of human ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7876496/]
- 73. Induced Pluripotent Stem Cells (iPSCs) Market Prospects ... [https://www.businesswire.com/news/home/20251111238841/en/Induced-Pluripotent-Stem-Cells-iPSCs-Market-Prospects-2025-2033-Industry-Gains-Traction-with-Rising-Investments-and-Collaborations---ResearchAndMarkets.com]